
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
Вернуться к номеру
Липоидозы
Авторы: И.Ю. Чибисова, Донецкий национальный медицинский университет им. М. Горького
Версия для печати
В настоящей статье рассматривается ряд наследственных ферментопатий, которые, ввиду своей относительно редкой встречаемости в общей популяции, зачастую не учитываются врачами при проведении дифференциальной диагностики. Ситуация усугубляется тем, что данная патология не является прерогативой врачей одной специальности, а может встречаться в практике гематологов, гастроэнтерологов, ревматологов, психоневрологов, невропатологов и особенно педиатров.
Обширный класс наследственных ферментопатий, или, иначе говоря, лизосомные болезни накопления (ЛБН), включает около 40 нозологических единиц и характеризуется генетической гетерогенностью и выраженным клиническим полиморфизмом. Практически все ЛБН имеют прогредиентное течение и в большинстве случаев приводят к ранней инвалидизации и преждевременной смерти. Лишь немногие формы ЛБН характеризуются близкой к норме продолжительностью жизни.
Наиболее типичными клиническими симптомами ЛБН являются поражение ЦНС, костные нарушения, гепатоспленомегалия, гематологические изменения. Клинические симптомы обусловлены преимущественным накоплением патологического материала в том или ином органе. Нарушение ферментных систем приводит к накоплению соответствующих специфических метаболитов в макрофагальных элементах с последующей инфильтрацией органов и тканей.
Наиболее часто из ЛБН в клинической практике встречается болезнь Гоше. Заболевание впервые описано P.E. Gaucher в
Патогенез многообразных клинических проявлений болезни прежде связывали с механическим накоплением клеток Гоше внутри органов и тканей и, как следствие, с нарушением их структуры и функции. Однако смущало несоответствие между относительно небольшим процентом патологических клеток в таких органах-мишенях, как костный мозг, кости, печень, и значительной степенью их разрушения (кости) и/или дисфункции (цитопении, цирроз печени). Успехи в изучении клеточной биологии и физиологии системы макрофагов (СМФ) дали возможность рассмотреть патофизиологию болезни Гоше с новых позиций.
СМФ — сложная саморегулирующаяся система, которая включает клетки-предшественники, локализующиеся в костном мозге; моноциты — пул относительно незрелых клеток, несколько часов циркулирующих в крови, и макрофаги — конечную стадию дифференцировки этой линии. В соответствии с локализацией различают органо- и тканеспецифические типы макрофагов. Наибольшее количество этих клеток содержится в селезенке и печени. Функции макрофагов исключительно многообразны и, помимо участия в обмене липидов, углеводов, метаболизме железа, включают клиренс крови и лимфы от токсинов, бактерий и других чужеродных агентов, регуляцию гемопоэза, гемостаза и фиброгенеза. Специфические функции макрофагов реализуются благодаря их способности к активации — физиологической реакции, которая вызывается действием различных факторов экзогенного (инфекция, антигены, токсины) и эндогенного происхождения. В случае болезни Гоше эндогенными стимулирующими факторами служат неутилизированные липиды, накапливающиеся в цитоплазме макрофагов. Их активация сопровождается секрецией широкого спектра биологически активных продуктов, в том числе провоспалительных цитокинов, цитотоксических и прокоагулянтных факторов. Общим результатом этого является каскад иммунопатологических реакций, лежащих в основе многих клинических симптомов и синдромов. Наиболее известны симптомы общей интоксикации (лихорадка и потеря веса), менее изучены изменения в системе крови и цитотоксические эффекты.
Наиболее выраженные гистологические изменения определяют в селезенке, которая достигает огромных размеров. На срезах пульпа имеет «мраморный» рисунок — множество желтоватых пятнышек, образованных скоплением клеток Гоше. Аналогичные инфильтраты находят в печени, которая значительно увеличивается и теряет дольчатую структуру. В костной ткани наблюдаются очаговые поражения с развитием деструкции кости, сочетающиеся со склеротическими изменениями. Процесс главным образом затрагивает трубчатые кости — берцовую, бедренную, плечевую. Отложение патологического субстрата происходит и в лимфатических узлах, что ведет к их умеренной гиперплазии. Также отмечаются отложения в коже с изменением ее окраски, особенно на открытых участках. Патологические изменения находят и в роговице глаза. В костном мозге в отличие от селезенки клетки Гоше рассеяны среди нормальных гемоцитов. Иногда специфические отложения находят и в легких. При хронической форме болезни Гоше круг пораженных органов обычно ограничен кроветворной, лимфатической и костной системами. При острой форме отмечается значительное накопление глюкоцереброзида в нервной системе, также поражаются глаза, пищеварительный тракт, надпочечники, почки. В нервной системе имеются большие скопления клеток Гоше вокруг сосудов. Развивается дегенерация нейронов таламуса, базальных ганглиев ядер ствола, спинного мозга, мозжечка.
Тяжесть клинических признаков при болезни Гоше находится в прямой зависимости от энзимного дефекта. Чем больше снижена активность фермента, тем в более раннем возрасте и в более тяжелой форме проявляется заболевание.
Выделяют три фенотипа болезни Гоше: 1-й тип — неневрологический — встречается как у детей, так и у взрослых, наиболее часто у евреев-ашкенази; 2-й тип — ювенильный неврологический — наиболее тяжелый, встречается у детей грудного возраста; 3-й тип — юношеский неврологический, характерными чертами которого являются, наряду с инфильтрацией органов ретикулоэндотелиальной системы, неврологическая симптоматика с поражением глазодвигательных нервов и выраженный полиморфизм неврологических проявлений.
При 1-м типе болезнь Гоше характеризуется хроническим и наиболее благоприятным течением. Заболевание может возникать в детском возрасте и долгое время протекать бессимптомно. Первые признаки появляются в подростковом или взрослом возрасте. В связи с длительным бессимптомным начальным периодом даже анамнестически бывает трудно установить момент возникновения заболевания. Первые симптомы, как правило, обусловлены поражением селезенки — возникают нерезкая боль в левом подреберье, чувство полноты в этой области. У детей описан вариант начала болезни Гоше с поражения костной системы, когда в клинической картине доминируют боль в костях ног, хромота. При этом варианте нервная система, как правило, не поражается и отставания в психофизическом развитии не наблюдается. В основном происходит инфильтрация селезенки, печени, скелета и костного мозга. Ведущий клинический симптом — спленомегалия. Селезенка плотная, бугристая, может достигать гигантских размеров, ее нижний полюс иногда определяется у входа в малый таз. Несколько позже в процесс вовлекается печень, которая также может достигать больших размеров, при пальпации плотная, безболезненная. Желтуха и асцит для болезни Гоше не характерны. Выраженная гепатоспленомегалия приводит к значительному увеличению объема живота. Больные имеют характерный вид — большой выпирающий живот (рис. 1).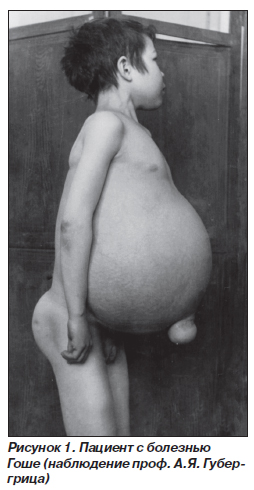 Часто развиваются костные изменения, беспокоит боль в трубчатых костях. Из-за интенсивной боли ограничена подвижность, частыми осложнениями являются остеопенические кризы, которые сопровождаются болевым синдромом, возможны спонтанные переломы трубчатых костей, остеомиелиты, деформация скелета, формирование кифосколиоза. При всех трех типах болезни вовлекается костная система, однако при 1-м типе болезни Гоше костная патология является основным инвалидизирующим фактором. При рентгенологическом исследовании определяют остеопороз и кисты костей, расширение костномозгового канала и атрофию кортикального слоя трубчатых костей. Характерные рентгенологические особенности определяют в бедренных и большеберцовых костях. Вследствие гиперостоза симметрично с обеих сторон диафизы кости имеют булавовидную деформацию — «колбу Эрленмейера». Кости черепа обычно не страдают. При данном варианте болезни Гоше наблюдаются кожные изменения в виде участков желто-коричневой пигментации с преимущественной локализацией на спине, кистях, но, как правило, они возникают в поздний период. Аналогичные изменения могут быть на слизистых оболочках. Увеличение лимфатических узлов — относительно редкий симптом и носит умеренный характер. Картина крови не имеет специфических признаков. Отмечаются умеренная анемия, лейкопения, тромбоцитопения, нарастающие в терминальный период. С одной стороны, панцитопения обусловлена инфильтрацией костного мозга клетками Гоше, с другой — гиперспленизмом. Геморрагический синдром длительное время носит умеренный характер и проявляется в виде подкожных кровоизлияний, позже присоединяются носовые и желудочно-кишечные кровотечения. Это обусловлено как тромбоцитопенией, так и развитием портальной гипертензии, сопровождающейся варикозным расширением вен пищеварительного тракта. Клетки Гоше в периферической крови не встречаются, их обнаруживают при исследовании миелограммы.
Часто развиваются костные изменения, беспокоит боль в трубчатых костях. Из-за интенсивной боли ограничена подвижность, частыми осложнениями являются остеопенические кризы, которые сопровождаются болевым синдромом, возможны спонтанные переломы трубчатых костей, остеомиелиты, деформация скелета, формирование кифосколиоза. При всех трех типах болезни вовлекается костная система, однако при 1-м типе болезни Гоше костная патология является основным инвалидизирующим фактором. При рентгенологическом исследовании определяют остеопороз и кисты костей, расширение костномозгового канала и атрофию кортикального слоя трубчатых костей. Характерные рентгенологические особенности определяют в бедренных и большеберцовых костях. Вследствие гиперостоза симметрично с обеих сторон диафизы кости имеют булавовидную деформацию — «колбу Эрленмейера». Кости черепа обычно не страдают. При данном варианте болезни Гоше наблюдаются кожные изменения в виде участков желто-коричневой пигментации с преимущественной локализацией на спине, кистях, но, как правило, они возникают в поздний период. Аналогичные изменения могут быть на слизистых оболочках. Увеличение лимфатических узлов — относительно редкий симптом и носит умеренный характер. Картина крови не имеет специфических признаков. Отмечаются умеренная анемия, лейкопения, тромбоцитопения, нарастающие в терминальный период. С одной стороны, панцитопения обусловлена инфильтрацией костного мозга клетками Гоше, с другой — гиперспленизмом. Геморрагический синдром длительное время носит умеренный характер и проявляется в виде подкожных кровоизлияний, позже присоединяются носовые и желудочно-кишечные кровотечения. Это обусловлено как тромбоцитопенией, так и развитием портальной гипертензии, сопровождающейся варикозным расширением вен пищеварительного тракта. Клетки Гоше в периферической крови не встречаются, их обнаруживают при исследовании миелограммы.
Общее состояние больных длительно может оставаться удовлетворительным. Отмечаются слабость, утомляемость, субфебрилитет. Долгое время сохраняется работоспособность. Отмечено, что существует прямая корреляционная зависимость между возрастом, в котором проявилась болезнь Гоше, и тяжестью заболевания.
2-й тип болезни Гоше характеризуется дебютом в грудном возрасте и острым злокачественным течением патологического процесса. Дети рождаются внешне здоровыми. Вскоре у ребенка появляются признаки отставания в психомоторном развитии, отмечается плохая прибавка в массе и росте. Отсутствует адекватная возрасту реакция на окружающее, дети вялые, апатичные. Острое течение характеризуется генерализованным поражением внутренних органов, что определяет разнообразие клинических симптомов. В связи с гепатоспленомегалией быстро увеличивается в размере живот. Характерны различные неврологические нарушения — задержка в психоневрологическом развитии, приступы тонико-клонических судорог, опистотонус. Бульбарные расстройства характеризуются «беззвучным криком» ребенка, поражением глазодвигательных нервов в виде сходящегося косоглазия, нарушением глотания. Развивается мышечная гипертония — ригидность затылочных мышц, согнутые невыпрямляющиеся конечности; спастическая ригидность может доходить до полной децеребрации. Умеренно увеличиваются лимфатические узлы. Специфическое поражение легких приводит к развитию дыхательных расстройств. В крови нарастает анемия и тромбоцитопения, развивается геморрагический синдром. Прогноз в грудном возрасте неблагоприятный. Нарушение глотания приводит к невозможности приема пищи, тяжелому истощению. Часто смерть наступает от вторичных осложнений — пневмонии, аспирации пищевых масс. Летальный исход наблюдается на первом году жизни, реже — на втором.
Острое течение также отмечается и при 3-м типе болезни Гоше — юношеском неврологическом — у детей дошкольного и младшего школьного возраста. Процесс также характеризуется значительной генерализацией, поражением различных внутренних органов, инфильтрацией костного мозга, но протекает менее бурно. Смерть наступает при явлениях дистрофии и тяжелых неврологических нарушениях. Длительность заболевания обычно не превышает двух лет от момента появления первых симптомов.
В детском возрасте диагностика болезни Гоше наиболее затруднительна, так как ведущий симптомокомплекс — гепатоспленомегалия, панцитопения — наблюдается при многих других заболеваниях. В связи с этим болезнь Гоше должна включаться в дифференциальную диагностику у детей и взрослых при неясной гепатоспленомегалии, измененных печеночных функциональных тестах, кровотечениях, изменениях в костной системе. Критерием достоверного диагноза является обнаружение клеток Гоше в пунктатах селезенки, печени, костного мозга.
Лечение болезни Гоше включает в себя несколько возможных вариантов:
В паллиативной терапии нуждаются все больные. Этот вид лечения направлен на коррекцию сопутствующих заболеваний и осложнений болезни Гоше. До недавнего времени считалось, что спленэктомия является основным методом лечения заболевания, однако годы наблюдения за пациентами показали, что удаление селезенки улучшает состояние больных на 1–2 года, затем в результате того, что утерян плацдарм для разрушения неактивных «нагруженных» макрофагов, патологические клетки депонируются в костной ткани, резко ухудшая костную патологию и приводя к прогрессированию заболевания. Поэтому вопрос об удалении селезенки может решаться только строго индивидуально и лишь при выраженных симптомах гиперспленизма. Вторым методом, применяемым в педиатрии, является терапия системного остеопороза. Это имеет и определенную патогенетическую направленность. Нередко встает вопрос об ортопедической коррекции костной патологии. Предпринимаются попытки лечения этой наследственной патологии путем пересадки костного мозга. Метод позволяет на 5–7 лет полностью стабилизировать состояние больного, однако имеет ряд существенных недостатков (поиск совместимого донора, повышенная вероятность реакции «трансплантат против хозяина», невозможность полного излечения).
Метод генной инженерии в настоящее время находится в стадии разработки. Таким образом, безальтернативным методом лечения пациентов с болезнью Гоше можно считать ферментозаместительную терапию.
Ферментозаместительная терапия проводится рекомбинантной глюкоцереброзидазой (церезим) по различным схемам. Уже через 3 месяца от начала терапии наблюдается положительная динамика со стороны паренхиматозных органов, показателей периферической крови. Больные дети перестают отставать в росте и весе.
К мероприятиям социального плана, направленным на профилактику функциональной перегрузки СМФ, следует отнести просветительную работу (с родителями и детьми), цель которой — разъяснить в доступной форме природу заболевания и необходимость вести здоровый образ жизни как единственную возможную меру индивидуальной профилактики функциональной перегрузки дефектного звена метаболизма. Основная задача — воспрепятствовать развитию вредных привычек и устранить дополнительную нагрузку на СМФ в виде привычных экзогенных интоксикаций (алкоголь, никотин) и недоброкачественных пищевых продуктов.
Следует особо подчеркнуть опасность необоснованной фармакотерапии, к которой прежде всего относятся препараты железа, назначаемые длительно по поводу гипохромной анемии. Исследование обмена железа у всех пациентов с болезнью Гоше показало отсутствие дефицита железа. Лечение ферропрепаратами во всех случаях сопровождалось усугублением исходной тромбоцитопении.
Реализация всех перечисленных пунктов профилактики и лечения болезни Гоше требует преемственности в работе педиатров и специалистов, работающих со взрослыми пациентами, организации диспансерного наблюдения за такими больными в специализированных центрах, которые располагают квалифицированным персоналом и обладают технической возможностью для диагностики, лечения и мониторинга пациентов с болезнью Гоше.
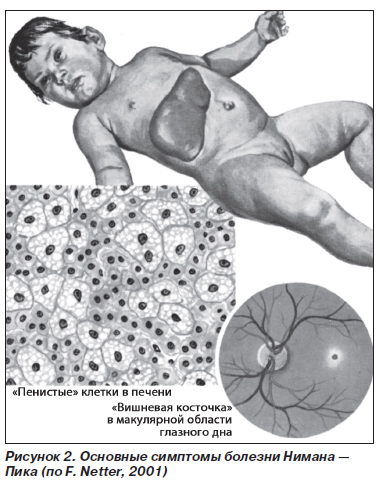
Болезнь Нимана — Пика (ретикулогистиоцитарный сфингомиелиноз, фосфатидный липидоз) (рис. 2) — наследственное заболевание, обусловленное накоплением в клетках ретикулогистиоцитарной системы фосфолипидов, преимущественно сфингомиелина и холестерина. Наследуется по рецессивному типу. В семьях с больными детьми часто имеются кровнородственные браки.
Заболевание впервые описано R. Niemann в 1914 г. и более подробно L. Pick в 1922 г. Встречается реже, чем болезнь Гоше. В основе патогенеза лежит дефицит фермента сфингомиелинидазы. При возникновении заболевания в грудном возрасте (от 1 до 2 месяцев) активность фермента почти полностью отсутствует, составляет 5–7 % таковой у здоровых людей. Энзимный дефект приводит к отложению сфингомиелина в селезенке, печени, лимфатических узлах, железах внутренней секреции. Патологический субстрат представлен макрофагальными элементами — клетками Нимана — Пика. Это специфические клетки, но могут встречаться и при некоторых других болезнях накопления.
Наиболее часто (80 % всех случаев) встречается острая форма заболевания, дебют которого приходится на грудной возраст. Дети рождаются внешне здоровыми, и период новорожденности обычно протекает нормально. Затем отмечаются потеря аппетита, снижение массы тела, нарастает дистрофия. Задерживается психомоторное развитие, ребенок перестает фиксировать взгляд, реагировать на окружающее. Лицо напоминает больных с синдромом Дауна — открытый рот, высунутый язык. Отмечаются умеренная лимфаденопатия, значительная гепатоспленомегалия. Органы плотные, безболезненные. Неврологический статус характеризуется развитием спастических парезов и параличей, движения некоординированы, сухожильные рефлексы сохранены. Позднее при развитии мышечной гипотонии они снижаются. В развернутой стадии развиваются моторные и психические нарушения, вплоть до идиотии. У детей появляются слепота и глухота. При офтальмоскопическом исследовании обнаруживают атрофию сосков зрительных нервов и вишнево-красное пятнышко в макулярной области глазного дна (симптом «вишневой косточки»). Кожа приобретает желто-коричневый оттенок. У детей повышается восприимчивость к инфекциям, довольно часто обнаруживаются лейкопения, гипохромная анемия как результат гиперспленизма. Количество тромбоцитов обычно нормальное. В грудном возрасте болезнь Нимана — Пика быстро прогрессирует, терминальная стадия сопровождается асцитом, легочно-сердечной недостаточностью в результате инфильтрации клетками Нимана — Пика бронхов и легких. Смерть наступает на 2-м году жизни ребенка.
Хроническая форма болезни Нимана — Пика встречается у детей старшего возраста и у взрослых. Ведущим клиническим синдромом является гепатоспленомегалия, возможны эндокринные нарушения. В отдельных случаях клиническая картина напоминает таковую при циррозе печени, фиброзе легких. У детей старшего возраста часто возникают неврологические нарушения в виде спастических парезов, приступов судорог, мозжечковых симптомов, расстройства походки, координации движений. Заболевание прогрессирует медленно, но неуклонно и заканчивается смертью больных.
Диагноз является достоверным при обнаружении клеток Нимана — Пика в пунктате селезенки, костного мозга, лимфатических узлов, печени. В настоящее время эффективного лечения не существует, применяется симптоматическая терапия. На стадии клинических испытаний находятся рекомбинантные препараты.
Болезнь Вольмана (генерализованный ксантоматоз) — тяжелая наследственная патология с аутосомно-рецессивным типом наследования, характеризуется накоплением холестеринэстеров в печени, селезенке, лимфатических узлах и надпочечниках вследствие недостаточности лизосомной холестеринэстергидролазы и, возможно, кислой липазы. Симптомы появляются в первые недели жизни ребенка: неукротимая рвота, диарея, дистрофия и обезвоживание, признаки поражения нервной системы — атаксия, деменция. Отмечается значительная гепатоспленомегалия, развивается ксантоматоз кожи. Для заболевания чрезвычайно характерно поражение надпочечников в виде кальцинации коры. Диагностика основывается на обнаружении типичных пенистых клеток в печени, селезенке и других органах. Прогноз неблагоприятный, дети умирают в первом полугодии жизни.
Болезнь накопления холестеринэстеров связана с врожденным лизосомным ферментным дефектом, по-видимому α-нафтилацетатэстеразной недостаточностью, которая приводит к накоплению холестеринэстеров в печени. В клинической картине доминирует гепатомегалия, уже в детском возрасте развивается цирроз печени с осложнениями. У большинства больных повышено содержание атерогенных липопротеидов. Диагностика основывается на выявлении пенистых клеток в перипортальных полях, исследовании липидов, изучении ферментов в культуре фибробластов. В отличие от болезни Вольмана кальцинации надпочечников не определяется. Прогноз зависит от особенностей течения цирроза печени. Специфическая терапия отсутствует.
Все вышеперечисленные заболевания являются довольно редкими и при проведении дифференциальной диагностики вызывают значительные трудности, однако в свете стремительного развития генной инженерии, когда появляется реальная возможность предотвратить раннюю инвалидизацию и смерть пациентов, незнание или невнимание к этой группе заболеваний является просто непростительным. В настоящее время возможно проведение заместительной терапии у пациентов с болезнью Гоше, и, как считают специалисты, в ближайшие 10 лет будут разработаны рекомбинантные препараты для лечения практически всех лизосомных болезней накопления.