
Газета «Новости медицины и фармации» 15(335) 2010
Вернуться к номеру
Инсулинорезистентность и канцерогенез
Авторы: Н.А. Кравчун, ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского АМН Украины»
Версия для печати
В настоящее время активно обсуждается проблема развития онкологических заболеваний у пациентов с сахарным диабетом (СД) 2го типа. Уже сформирована убедительная точка зрения относительно возрастающего онкологического риска при метаболическом синдроме (МС). На сегодняшний день признана роль инсулинорезистентности (ИР), локального эстрогенообразования и гиперандрогенизации в предрасположенности к развитию некоторых онкологических заболеваний [1, 2]. Также доказано значение молекулярногенетических факторов, частично ассоциированных с гормональнометаболическими нарушениями, как основы формирования различных вариантов опухолей [3]. Общность патологических механизмов канцерогенеза и МС, СД 2го типа позволяет выделить дополнительные факторы риска возникновения и развития ракового перерождения тканей, а также наметить возможные пути немедикаментозной и медикаментозной их профилактики.
В 1997 г. была показана взаимосвязь между ИР и развитием рака молочной железы и толстого кишечника. Это исследование было проведено на очень большом количестве пациентов — 22 561 мужчина и 18 495 женщин — в течение 7 лет. Авторами было установлено, что у 27 % женщин, умерших от рака толстого кишечника, имелась ИР (группа контроля — 3 %). Среди обследованных пациентов у 9,5 % женщин, умерших от рака молочной железы, также была диагностирована ИР в сочетании с гиперандрогенией (группа контроля — 3,5 %) [4].
В исследовании, проведенном финскими учеными, при более чем 10летнем наблюдении за мужчинами с МС рак простаты обнаруживался в 1,9 раза чаще по сравнению с лицами без МС.
D.C. Malins и соавт. в 1998 г. показали, что у женщин с ИР и высоким уровнем инсулиноподобных факторов роста (ИФР) карцинома молочной железы возрастает в 2–3 раза [5].
Среди больных с гистологически верифицированным раком эндометрия, находившихся в СанктПетербургском городском онкологическом диспансере, на протяжении 3 лет признаки ИР были диагностированы у 35 %.
Данные крупномасштабных исследований Американского общества рака (2005 г.) свидетельствуют о том, что ожирение является причиной злокачественных новообразований у 14 % мужчин и 20 % женщин. При этом у мужчин риск умереть от рака возрастает на 52 %, а у женщин — на 62 % по сравнению с лицами аналогичного пола и возраста без ожирения.
В исследовании, в котором приняло участие около 900 тыс. человек, было показано, что среди женщин с избыточной массой тела и ожирением рак матки возникал в 6 раз чаще, шейки матки — в 3 раза, рак почек — в 5 раз, молочной железы, желчного пузыря, поджелудочной железы и пищевода — в 2 раза чаще по сравнению с лицами с нормальной массой тела. Среди мужчин с ожирением по сравнению с лицами с нормальной массой тела рак печени диагностировался в 6 раз чаще, поджелудочной железы — в 2 раза, желчного пузыря, желудка и прямой кишки — на 75 % чаще.
Результаты исследования итальянских ученых (Институт фармакологических исследований в Милане) свидетельствуют о достоверной прямой связи между гиперхолестеринемией и развитием рака предстательной железы.
О том, что ИР и гиперинсулинемия сопряжены с риском рака поджелудочной железы у мужчин, свидетельствуют данные исследования, проведенного американскими учеными (2005 г.). В данной работе задействовано 30 000 участников, 400 здоровых добровольцев в группе контроля; длительность наблюдения — 17 лет. Авторы делают вывод о том, что СД 2го типа является фактором риска рака поджелудочной железы, что связывают с активностью инсулиноподобного фактора роста [1].
По исследованиям, проведенным в эксперименте, инсулин действительно способствует росту раковых клеток. К митогенным (ростстимулирующим) относят долгосрочные эффекты инсулина, реализуемые на генетическом уровне за счет индукции экспрессии ряда специфических генов, стимуляции синтеза ДНК и белков, ведущих в конечном счете к усилению клеточного роста [6–8].
Описано митогенное действие инсулина, которое реализуется через сложную цепь сигнальных событий: активация цАМФзависимой протеинкиназы, фосфорилирование и активирование фактора транскрипции в ядре, индукция экспрессии цАМФзависимых генов, что в итоге приводит к стимуляции клеточной пролиферации [9]. Не исключено, что представленный митогенный путь инсулина находится во взаимодействии с основным митогенным сигнальным путем организма, включающим каскад митогенактивируемых протеинкиназ, запускаемых через рецептор тирозинкиназного типа.
При выяснении механизмов действия инсулина на экспрессию генов, ответственных за клеточную пролиферацию и утилизацию глюкозы, было установлено, что регуляция инсулином генов EIF1 (early growth response gene) и глицеральдегид3фосфатдегидрогеназы (a metabolic response gene) опосредуется через различные сигнальные пути [10]. По мнению ряда авторов, влияние инсулина на клеточный рост связано с активацией протеинкиназ и стимуляцией процесса фосфорилирования белков, метаболическое же действие реализуется за счет ингибирования этого процесса, а также усиления реакции дефосфорилирования эффекторных белков, вызванного активацией протеинфосфатаз [11–13]. Интересен такой парадоксальный факт: инсулин регулирует степень фосфорилирования белков по тирозину и серину/треонину, стимулируя фосфорилирование одних белков и в то же время дефосфорилирование других. Дефосфорилирование, индуцируемое инсулином, связано с рядом его метаболических эффектов [14].
Представленные исследования свидетельствуют о наличии митогенных и метаболических сигнальных путей действия инсулина в клетке.
Инсулин также стимулирует пролиферацию некоторых клеток в культуре. При изучении регуляции роста в основном используются культуры фибробластов. Инсулин в таких клетках усиливает способность фактора роста фибробластов, тромбоцитарного фактора роста, фактора роста эпидермиса, стимулирующих рост опухолей форболовых эфиров, простагландина F2a, вазопрессина, аналогов сАМР активировать размножение клеток после удаления их из среды сыворотки.
Необходимо подчеркнуть, что биохимический механизм влияния инсулина на репликацию клеток на сегодняшний день до конца не выяснен. Предполагается, что он основан на анаболическом действии гормона.
Не исключено, что в этом процессе играет роль влияние на поглощение глюкозы, фосфата, нейтральных аминокислот и катионов [15]. Инсулин может стимулировать репликацию благодаря своей способности активировать или инактивировать ферменты путем регуляции скорости и степени фосфорилирования белков или регулируя синтез ферментов. Даже очень низкие концентрации инсулина стимулируют репликацию (скорее всего, через инсулиновый рецептор), причем чаще всего это происходит в отсутствие других пептидных факторов роста.
Инсулин является обязательным компонентом всех известных средств для культивирования тканей, поддерживает рост и репликацию множества клеток эпителиального происхождения, в частности гепатоцитов, клеток гепатомы, клеток опухолей коры надпочечников, клеток карциномы молочной железы.
Инсулиновый рецептор, как и рецепторы других факторов роста, обладает тирозинкиназной активностью [16, 17]. Необходимо подчеркнуть, что 10 онкогенных продуктов, многие из которых участвуют в стимуляции репликации злокачественных клеток, также представляют собой тирозинкиназы.
Необходимо обратить внимание на антиапоптотическое действие инсулина. О его наличии можно судить, рассмотрев взаимосвязь двух антагонистических процессов: клеточного обновления и клеточной гибели, которые согласованно регулируются для достижения соответствующей ситуации клеточного ответа. Так, установлено, что инсулин и ИФР1 играют ключевую роль в стимуляции развития, роста и органогенеза, которая осуществляется через митогенное и антиапоптотическое действие [18].
СД 2го типа является фактором риска и демонстрирует положительную корреляцию с раком прямой кишки.
В исследованиях AlphaTocopherol, BetaCarotene Cancer Prevention Study участвовало 28 983 курящих мужчины, у которых в период с 1985 по 2002 г. был диагностирован рак прямой кишки или параректальный рак. Лица, имеющие 3 проявления синдрома инсулинорезистентности (гипертензия, ИМТ > > 25 кг/м2, холестерин ЛПВП < 40 мг/дл (< 1,55 ммоль/л)), имеют статистически значимое повышение риска параректального рака. Полученные результаты говорят в пользу предположения о том, что значительная корреляция, наблюдаемая между метаболическими нарушениями, связанными с инсулинорезистентностью, и параректальным раком, определяется в основном ожирением [19].
В исследование «Диабет и риск рака поджелудочной железы» было включено 720 пациентов с раком поджелудочной железы и 720 контрольных пациентов из 14 итальянских исследовательских центров. 164 пациента (22,8 %) с раком и 60 (8,3 %) человек из группы контроля страдали СД.
У 49 больных (2 %) СД был диагностирован параллельно с раком. Корреляция между этими заболеваниями была достоверно значима (частота вероятности 3,04; 95% интервал достоверности от 2,21 до 4,17). Однако у пациентов со стажем СД 3 года и более эта достоверность заметно снижалась (частота вероятности 1,43; 95% интервал достоверности от 0,98 до 2,07). Все пациенты с раком поджелудочной железы, кому диагноз «диабет» был поставлен до онкологического, имели инсулинонезависимую форму заболевания; в контрольной группе все пациенты, кроме одного, также имели инсулинонезависимый диабет. Авторы делают вывод, что диабет часто возникает у пациентов с раком поджелудочной железы и предположительно является следствием развития опухоли. Однако СД не является фактором риска онкопатологии поджелудочной железы [20].
В другом итальянском исследовании была изучена взаимосвязь диабета и рака эндометрия. Участие приняли 752 женщины в возрасте до 75 лет с гистологически подтвержденным раком эндометрия, из которых 132 больные (17,6 %) страдали СД. Контрольную группу составили 2606 пациенток, среди которых СД имелся у 116. Частота возникновения рака эндометрия у женщин с СД и ИМТ ниже 25 составила 3,0; с ИМТ от 25 до 29 — 3,6; с ИМТ 30 и более — 3,3. Учеными сделан вывод о взаимосвязи СД 2го типа, инсулинонезависимого, с раком эндометрия. Эта взаимосвязь может быть опосредована через снижение уровня эстрогенов, гиперинсулинемию или инсулиноподобный фактор роста1 [21].
Приведенные литературные данные свидетельствуют, что инсулин формирует митогенную и метаболическую основу для предотвращения опухолевого роста. В этом ключе значительна роль гиперинсулинемии как самостоятельного фактора риска в развитии опухолей ряда органов. Избыточная выработка инсулина может модифицировать и стероидогенез за счет как прямого влияния на синтез стероидов в яичниках, так и опосредованного путем угнетения в печени продукции глобулина, связывающего половые гормоны [22, 23].
Обращает на себя внимание также значительно большая распространенность среди больных СД 2го типа с гиперинсулинемией злокачественных новообразований.
Возможные механизмы развития канцерогенеза в условиях ИР могут быть представлены схематически (рис. 1).
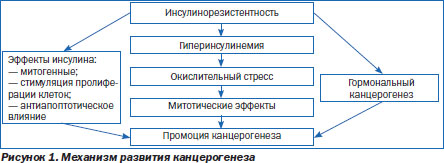
Каков медикаментозный путь положительного воздействия не только на уровень глюкозы и нормализацию уровня инсулина в крови, но и на снижение митотической активности инсулина?
В этом направлении на сегодняшний день проведено достаточное количество исследований.
Так, изучены эффекты антидиабетического препарата метформина на рост опухолей. Было показано, что прием метформина способствует уменьшению роста опухолей [24].
Исследованы эффекты метформина на раковые клетки простаты in vitro и in vivo. Показано, что метформин подавляет пролиферацию DU145, PC3 и LNCaP раковых клеток в 50 % случаев, снижает жизнеспособность клеток и оказывает незначительные эффекты на эпителиальные клетки простаты линии Р69. Авторами установлено, что метформин не индуцирует апоптоз, но блокирует клеточный цикл в фазе G0G1. Эта блокада сопровождается значительным снижением уровня циклического протеина D1, что обусловливает антинеопластические эффекты метформина и может быть использовано в эпидемиологических исследованиях [25].
Сотрудниками Университета Небраска Медицинского центра из Омахи (США) проведено экспериментальное исследование по предотвращению развития рака поджелудочной железы у хомячков в результате приема метформина. Были взяты 2 группы толстых хомячков. Первая группа получала метформин перорально с питьем, вторая группа была контрольной. Обе группы подвергали воздействию панкреатического канцерогена Nнитрособисамина. Несмотря на то что у 50 % животных были обнаружены опухоли, ни одно из них не относилось к исследуемой группе, принимавшей метформин. Интересно, что больше гиперплазированных и предраковых клеток (особенно панкреацитов) было обнаружено именно во второй группе. Таким образом, авторы делают заключение о взаимосвязи рака поджелудочной железы и ожирения, ассоциированного, как правило, с инсулинорезистентностью, и способности метформина предотвращать развитие опухолей [26–28].
Показано также наличие антинеопластической активности метформина у онкологических пациентов с гиперинсулинемией. На культуре клеток LLC1 карциномы легких линии мышей C57BL/6J продемонстрировано, что метформин через увеличение активации рецепторов инсулина и увеличение фосфорилирования AMPкиназы способствует снижению неопластической пролиферации клеток [29].
Учеными из Университета МакГилла (Монреаль, Канада) сделано заключение, что метформин — препарат, широко применяемый в лечении СД 2го типа, — способен снижать риск возникновения рака и улучшать онкологический прогноз. Исследовались эффекты метформина на раковые эпителиальные клетки яичников путем определения цитотоксичности. Авторами сделан вывод, что метформин значительно ингибирует рост раковых клеток и усиливает действие цисплатина. Доклинические исследования свидетельствуют об эффективности использования метформина при раке яичников [30].
Известно, что метформин действует через активацию АМРК (АМРзависимую киназу) [31]. АМРК является регулятором клеточного энергетического гомеостаза, особенно в условиях стресса. Когда метформин активирует АМРК, следует значительное подавление клеточной пролиферации и активация АМРК 1/2 в эстрогеновых рецепторах a (ERa) как негативных (MDAMB231, MDAMB435), так и позитивных (MCF7, T47D) клеточных линий рака молочной железы человека. Лечение метформином показало увеличение экспрессии васкулярного эндотелиального фактора роста внутри опухоли и уменьшение некроза. Однако лечение метформином было обосновано, так как он угнетает ИПФР1 и пролиферацию опухолевых клеток. Далее исследователи показали, что метформин значительно подавляет рост раковых клеток молочной железы in vitro, что связано с его воздействием на эстрогеновые рецепторы негативных клеток рака молочной железы [32].
Нами были проанализированы данные 194 историй болезни пациентов с СД 2го типа, находившихся в клинике ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского АМН Украины», за период с 2007 по 2009 гг. В качестве антидиабетической терапии они принимали метформин в средней суточной дозе 1000 мг в среднем в течение 5 лет. Длительность СД составила 9 ± 5 лет, средний возраст пациентов — 54 ± 10 лет. Среди пациентов этой группы не было отмечено ни одного случая онкопатологии.
Таким образом, представленный анализ литературы и собственные данные свидетельствуют о частом сочетании СД 2го типа и онкопатологии. Взаимосвязь инсулинорезистентности и развития онкопатологии сомнений не вызывает. Вместе с тем обнадеживают имеющиеся убедительные доказательства того, что регулярный и длительный прием метформина способствует профилактике онкопатологии у пациентов с СД 2го типа.
1. Метаболический синдром / Под ред. чл.корр. РАМН Г.Е. Ройтберга. — М.: МЕДпрессинформ, 2007. — 224 с.: илл.
2. Van Zwieten P.A., Mancia G. The metabolic syndrome — a therapeutic challenge: monograph. — Amsterdam: Van Zuiden Communications B.V. 2007. — 99 p.
3. Краевский Н.А., Смольянников А.В., Саркисов Д.С. Патологоанатомическая диагностика опухолей человека. — Медицина, 1993. — 60 с.
4. Link Shown Between Insulin Resistance, Colon And Breast Cancer [Electronic resource]. — Access mode : http://www.pslgroup.com.
5. Malins D.С., Polisaar N.L., Schaefer S. et al. A unified theory of carcinogenesis based on orderdisorder transitions in DNA structure as studied in the human ovary and breast // Proc. Nat. Acad. Sci. (USA). — 1998. — Vol. 95. — P. 76377642.
6. Gupta В.В.Р. et al. Mechanism of insulin action // Curr. Science. — 1997. — V. 73. — P. 9931003.
7. O’Brien R.M., Granner D.K. Regulation of gene expression by insulin // Physiol. Rev. — 1996. — V. 76. — P. 110961.
8. While M.F., Kanh C.R. The insulin signalling system // J. Biol. Chem. — 1994. — V. 269. — P. 14.
9. Перцева М.Н. Вклад эволюционной эндокринологии в изучение структуры и механизмов действия инсулина // Ж. эвол. биохим. и физиол. — 1999. — № 35. — С. 175186.
10. AlexanderBridges M., Buggs C., Giere L. et al. Models of insulin action on metabolic and growth response genes // Mol. Cell. Biochem. — 1992. — V. 109. — Р. 99105.
11. Saltiel A.R. Diversy signalling patways in the cellular actions of insulin // Am. J. Physiol. — 1996. — V. 270. — E375E385.
12. Lazar D.F., Wiese R.J., Brady M.J. et al. Mitogenactivated protein kinase inhibition does not block the stimulation of glucose utilization by insulin // J. Biol. Chem. — 1995. — V. 270. — Р. 20801807.
13. Cohen P. Dissection of the protein phosphorylation cascades involved in insulin and growth factor action // Biochem. Soc. Transact. — 1993. — V. 21. — P. 555567.
14. Бутрова С.А. Метаболический синдром: патогенез, клиника, диагностика, подходы к лечению // Рус. мед. журн. — 2001. — Т. 9. — С. 5660.
15. Благосклонная Я.В., Красильникова И.Е., Бабенко Ю.А. Ожирение и его потенциальная роль в развитии метаболического синдрома // Новые СанктПетербургские врачебные ведомости. — 1998. — № 4. — С. 4348.
16. Алмазов В.А., Благосклонная Я.В., Шляхто Е.В. и др. Метаболический сердечнососудистый синдром. — СПб.: СПбГМУ, 1999. — 208 с.
17. Благосклонная Я.В., Шляхто Е.В., Красильникова Е.И. Метаболический сердечнососудистый синдром // Рус. мед. журн. — 2001. — Т. 9, № 2. — С. 6771.
18. Butler A.A., Yakar S., Gevolb J.H. et al. Insulin like growth factorI receptor signal transduction: at the interface between physiology and cell biology // Comp. Biochem. Physiol. — 1998. — V. 121. — P. 1926.
19. Bowers K.A., Albanes D., Limburg P. et al. Prospective Study of Anthropometric and Clinical Measurements Associated with Insulin Resistance Syndrome and Colorectal Cancer in Male Smokers // Am. J. Epidemiol. — 2006. — Vol. 164, № 7. — P. 652664.
20. Gullo L., Pezzilli R., MorselliLabate A.M. Diabetes and the Risk of Pancreatic Cancer // The new England journal of medicine. — 1994. — Vol. 331, № 2. — P. 8184.
21. Parazzini F., Vecchia La C., Negri E. et al. Diabetes and endometrial cancer: an italian casecontrol study // Int. J. Cancer. — 1999. — Vol. 81. — P. 539542.
22. Bernstein L., Ross R.K., Henderson B.E. Cancer prevention: hormones // CancenPrinciples and practice of oncology. — 5th ed. / Еd. by V.T. DeVita et al. — Philadelphia: LippincottRaven Publ., 1997. — P. 677693.
23. Poretzky L., Cataldo N.A., Rosenwaks Z., Giiidice L.C. The insulinrelated ovarian regulatory system in health and disease // Endocrine Rev. — 1999. — Vol. 20. — P. 535582.
24. Buzzai M., Jones R.G., Amaravadi R.K. et al. Systemic Treatment with the Antidiabetic Drug Metformin Selectively Impairs p53Deficient Tumor Cell Growth // Cancer Res. — 2007. — Vol. 67, № 14. — P. 674552.
25. Ben Sahra I., Laurent K., Loubat A. et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level // Oncogene. — 2008. — Vol. 27. — P. 357686.
26. Schneider M.B., Matsuzaki H., Haorah J. et al. Prevention of pancreatic cancer induction in hamsters by metformin // Gastroenterology. — 2001. — Vol. 120, № 5. — P. 126370.
27. Дедов И.И., Бутрова С.А., Мищенко Б.П., Дзгоева Ф.Х. Применение метформина (сиофора) у больных с абдоминальным типом ожирения // Пробл. эндокринол. — 2000. — Т. 46, № 5. — С. 2529.
28. Шубина А.Т., Демидова И.Ю., Чернова Н.А., Карпов Ю.А. Метаболический синдром: возможности применения метформина // Рус. мед. журн. — 2001. — Т. 9, № 2. — С. 7781.
29. Algire C., Zakikhani M., Blouin M.J. et al. Metformin attenuates the stimulatory effect of a highenergy diet on in vivo LLC1 carcinoma growth // Endocr. Relat. Cancer. — 2008. — Vol. 15, № 3. — P. 833839.
30. Gotlieb W.H., Saumet J., Beauchamp M.C. et al. In vitro metformin antineoplastic activity in epithelial ovarian cancer // Gynecol. Oncol. — 2008. — Vol. 110, № 2. — P. 246250.
31. Старостина Е.Г. Бигуаниды: второе рождение // Новый мед. журн. — 1998. — № 1. — С. 28.
32. Phoenix K.N., Vumbaca F., Claffey K.P. Therapeutic Metformin/AMPK Activation Promotes the Angiogenic Phenotype in the ERa Negative MDAMB435 Breast Cancer Model // Breast Cancer Res. Treat. — 2009. — Vol. 113, № 1. — P. 101111.