
Международный неврологический журнал 3 (41) 2011
Вернуться к номеру
Болезнь Галлервордена — Шпатца (Клинический обзор и собственное наблюдение)
Авторы: Пономарев В.В., 5-я клиническая больница, г. Минск, Республика Беларусь
Рубрики: Неврология
Версия для печати
Представлен клинический разбор редкого аутосомно-рецессивного нейродегенеративного заболевания — болезни Галлервордена — Шпатца, начавшейся во взрослом возрасте. Диагноз подтвержден характерной клинической картиной и типичным МРТ-признаком в виде «глаза тигра». Обнаружена необычная диссоциация синдрома паркинсонизма в виде очевидного преобладания постуральной неустойчивости над слабой выраженностью гипокинезии и ригидности, а также отсутствие тремора и любых форм дистонии. Выявлен нетипичный для взрослой формы быстрый темп прогрессирования. Обсуждаются вопросы патоморфологии, патогенеза, классификации, дифференциальной диагностики, полиморфизма клинических проявлений, диагностических и лечебных подходов.
 Болезнь Галлервордена — Шпатца (БГШ) — наследственное дегенеративное заболевание нервной системы, связанное с накоплением железа в базальных ганглиях [1–4, 8]. Впервые описана немецкими морфологами Julius Hallervorden и Hugo Spatz в 1922 г. БГШ считается редким заболеванием, хотя истинная его частота остается неизвестной. Установлено наследование этой патологии по аутосомно-рецессивному типу. В клинической практике болезнь встречается в виде как спорадических, так и семейных случаев [4]. Выделено три типа в зависимости от возраста начала болезни: детский, подростковый и взрослый, а также ее типичный и атипичный варианты [10]. Значительный прорыв знаний в отношении механизмов развития болезни произошел в 2001 году, когда Zhou et al. впервые выявили дефект в гене, расположенном на коротком плече 20-й хромосомы, ответственный за синтез фермента пантотенаткиназы, после чего заболевание было переименовано в Pantothenate Kinase associated neurodegeneration (PKAN) [5, 7, 14]. Отчасти изменение термина также было связано с публикациями в литературе об участии Hallervorden и Spatz в программе «эвтаназии» психических больных, проводимой нацистами в годы войны [13].
Болезнь Галлервордена — Шпатца (БГШ) — наследственное дегенеративное заболевание нервной системы, связанное с накоплением железа в базальных ганглиях [1–4, 8]. Впервые описана немецкими морфологами Julius Hallervorden и Hugo Spatz в 1922 г. БГШ считается редким заболеванием, хотя истинная его частота остается неизвестной. Установлено наследование этой патологии по аутосомно-рецессивному типу. В клинической практике болезнь встречается в виде как спорадических, так и семейных случаев [4]. Выделено три типа в зависимости от возраста начала болезни: детский, подростковый и взрослый, а также ее типичный и атипичный варианты [10]. Значительный прорыв знаний в отношении механизмов развития болезни произошел в 2001 году, когда Zhou et al. впервые выявили дефект в гене, расположенном на коротком плече 20-й хромосомы, ответственный за синтез фермента пантотенаткиназы, после чего заболевание было переименовано в Pantothenate Kinase associated neurodegeneration (PKAN) [5, 7, 14]. Отчасти изменение термина также было связано с публикациями в литературе об участии Hallervorden и Spatz в программе «эвтаназии» психических больных, проводимой нацистами в годы войны [13].
Клинические проявления БГШ за 90 лет изучены достаточно подробно и отличаются значительным полиморфизмом симптомов. Наиболее характерными признаками заболевания считают синдром паркинсонизма, различные виды гиперкинезов, пирамидные знаки, снижение когнитивных функций, пигментную ретинопатию, атрофию зрительных нервов и др. [1, 2, 10]. Многие годы БГШ выявляли только посмертно. Однако в связи с широким внедрением в повседневную практику метода магнитно-резонансной томографии (МРТ) появилась возможность прижизненной диагностики этой патологии. Характерным МРТ-паттерном при БГШ принято считать овальную симметричную гиперинтенсивную зону в области бледного шара внутри более обширной гипоинтенсивной зоны. Этот типичный симптом БГШ назван «глазом тигра», и его формирование связано с внеклеточным накоплением железа в базальных ганглиях [9, 14, 15].
В русскоязычной литературе встречаются единичные публикации, посвященные этой крайне редкой патологии, в которых подчеркивается сложность ее диагностики как у детей, так и у взрослых [2, 4]. В связи с этим мы решили поделиться собственным опытом диагностики единичного клинического случая БГШ с поздним началом, сопроводив его анализом современной литературы.
Больной В., 30 лет, постоянный житель Республики Беларусь, впервые поступил во II неврологическое отделение 5-й клинической больницы г. Минска в 2003 году с жалобами на скованность и отсутствие плавности движений, нарушение равновесия, частые падения при резких поворотах. Болен 6 месяцев, когда без видимой причины появились и стали нарастать вышеописанные жалобы. Лечился по месту жительства с различными диагнозами (последний раз по поводу мультисистемной атрофии) без эффекта. Из перенесенных заболеваний отмечает простудные (редко) и легкую черепно-мозговую травму в 1998 г. Семейно-наследственный анамнез достоверно не отягощен. Объективно при поступлении общее состояние удовлетворительное, хорошо развита мышечная система. АД 120/80 мм рт.ст., пульс 70 уд/мин, ритмичный. Соматической патологии не выявлено. Неврологически: в сознании, ориентирован в месте и времени, слегка эйфоричен, незначительно снижена память на текущие события, ослаблено внимание. Черепные нервы без патологии, гипомимичен. Сила в конечностях достаточная, мышечный тонус диффузно повышен по экстрапирамидному типу. Сухожильно-периостальные рефлексы оживлены, S > D, брюшные, подошвенные равномерно ослаблены, патологических знаков нет. Чувствительность не нарушена. Координаторные пробы выполняет удовлетворительно, в позе Ромберга устойчив. Отмечена выраженная постуральная неустойчивость. Проба Тавенарда резко положительная: при ее проведении пациент падает назад без малейшей попытки удержать равновесие. Вегетативные тесты (ортостатический, синокаротидный, глазосердечный, солярный) были отрицательными. Функцию тазовых органов контролирует. Походка лишена плавности, отсутствуют физиологические синкинезии, при резких разворотах пациент часто теряет равновесие и падает из-за про- и латеропульсии. При обследовании: рутинные общеклинические анализы крови и мочи, биохимический анализ крови без патологии. Содержание церулоплазмина в сыворотке крови 1,7 ммоль/л (норма 1,3–3,5 ммоль/л), меди — 16,3 ммоль/л (норма 11–22 ммоль/л), железа — 15,7 ммоль/л (норма 9,5–30 ммоль/л). При нейропсихологическом тестировании общая оценка когнитивных функций по данным краткой шкалы оценки психического статуса (MMSE) не нарушена — 27 баллов (норма 28–30 баллов). Результаты выполнения теста «батарея лобной дисфункции» — 14 баллов (норма 18 баллов). Консультация окулиста: острота зрения 1,0, глазное дно без патологии, колец Кайзера — Флейшера не выявлено. МРТ головного мозга: базальные цистерны, желудочки мозга кортикальные борозды обычных размеров, релаксационные характеристики вещества мозга в норме. В проекции базальных ядер с обеих сторон определяются обширные симметричные гиперинтенсивные зоны с очагом просветления («глаз тигра», рис. 1). В целях дифференциальной диагностики этиологии поражения базальных ядер (болезнь Фара, гиперпаратиреоз) проведено исследование уровня паратгормона. Он оказался в пределах нормы. Уровни лактатата и пирувата в крови также были в пределах нормы. Получал леводопа-содержащие препараты, нейропротекторы, антиоксиданты без существенного эффекта. При катамнестическом наблюдении в течение 7 лет состояние больного продолжает медленно ухудшаться, перестал самостоятельно ходить из-за невозможности удержать равновесие. В последние годы передвигается при помощи коляски, память нарушена незначительно, движения в конечностях сохранены.
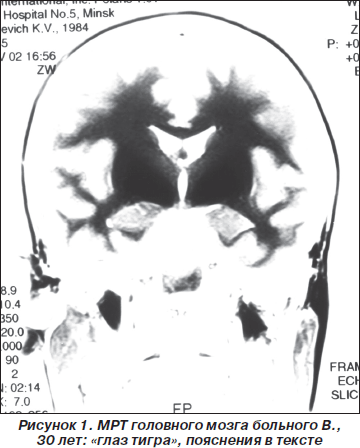
Таким образом, в представленном наблюдении заболевание началось в возрасте 29 лет, носило прогрессирующий характер и характеризовалось сочетанием следующих неврологических проявлений:
1) синдром паркинсонизма в виде легкой симметричной гипокинезии и экстрапирамидной ригидности, а также выраженной постуральной неустойчивости, которая превалировала, оставаясь ведущим проявлением на протяжении всей болезни;
2) легкая пирамидная недостаточность в форме латерализованного оживления сухожильно-периостальных и ослабления кожных рефлексов;
3) легкие когнитивные нарушения преимущественно подкоркового типа, что проявилось снижением показателя FAB при нормальных результатах MMSE.
Преобладание в клинической картине экстрапирамидного синдрома у пациента молодого возраста обосновывало проведение дифференциальной диагностики прежде всего с болезнью Паркинсона с ранним началом, болезнью Вильсона — Коновалова, а также со всеми формами вторичного паркинсонизма и другими дегенеративными заболеваниями ЦНС.
Болезнь Паркинсона с ранним началом (до 40 лет) проявляется изолированным экстрапирамидным синдромом, однако его отличительными особенностями являются асимметричный характер, частое наличие тремора покоя и, как правило, высокая чувствительность к препаратам леводопы, особенно в дебюте. Постуральная неустойчивость и когнитивные нарушения при этом заболевании появляются только при длительном течении, а пирамидные знаки и очаговые изменения на МРТ головного мозга не характерны.
Болезнь Вильсона — Коновалова (гепатоцеребральная дегенерация) при славянском генотипе заболевания также характеризуется началом до 30 лет и ведущими неврологическими нарушениями в виде дофарезистентного синдрома паркинсонизма. Однако эта патология отличается частым клиническим или субклиническим поражением печени и селезенки, формированием на радужной оболочке глаз колец Кайзера — Флейшера, типичными лабораторными нарушениями в виде снижения в сыворотке церулоплазмина и высокого содержания меди в крови и моче.
Все виды вторичного паркинсонизма (сосудистого, лекарственного, токсического, посттравматического, постэнцефалитического, постаноксического, гидроцефального) представилась возможность исключить на основании отсутствия специфических анамнестических сведений и наличия характерных изменений на МРТ головного мозга в виде «глаза тигра».
Среди прочих дегенеративных заболеваний по клиническим проявлениям к рассмотренному случаю были наиболее близки мультисистемная атрофия (пациент даже наблюдался по этому поводу) и нейроакантоцитоз. Однако в обоих случаях заболевания начинаются в более позднем возрасте (после 60 лет). Кардинальными отличительными признаками мультисистемной атрофии являются наличие в клинической картине прогрессирующей вегетативной недостаточности и более быстрый темп прогрессирования. Отсутствие наследственного анамнеза, указывающего на аутосомно-доминантный тип наследования, характерных симптомов сенсорной полиневропатии и типичных гематологических проявлений (акантоцитов или измененных эритроцитов) позволило отказаться от диагноза нейроакантоцитоза.
Таким образом, сочетание синдрома паркинсонизма, пирамидной симптоматики и нейропсихологических нарушений, постепенное начало, прогрессирующее течение, характерные изменения на МРТ в виде «глаза тигра» после проведения дифференциальной диагностики явились основанием для постановки диагноза БГШ, взрослая форма, с выраженным экстрапирамидным синдромом, пирамидной недостаточностью, легкими когнитивными нарушениями подкоркового типа.
Этиология заболевания неизвестна. Его патогенез связан с множественными (более 50) мутациями гена пантотенаткиназы (PKAN), расположенного на коротком плече 20-й хромосомы в локусе 20р12.3–р13 [4]. PKAN является регуляторным ферментом биосинтеза коэнзима А, катализирующего фосфорилирование пантотената, N-пантотенилцистеина и пантина [15]. Патологический ген приводит к снижению выработки данного фермента, что сопровождается избыточным накоплением цистеина в базальных ганглиях. Цистеин, в свою очередь, связывает ионы железа, формируя устойчивые комплексы, разрушающие нейрональные протеины [7]. В результате индуцируются реакции перекисного окисления с образованием свободных радикалов, также способствуя апоптозу нейронов базальных ганглиев. На месте разрушенных нейронов развивается глиоз, придающий пораженным тканям мозга губчатый вид [1]. Особенностью патогенеза БГШ является тот факт, что общий обмен железа при данном заболевании не нарушен [5]. Об этом свидетельствуют полученные нами данные о нормальном уровне железа в сыворотке крови в представленном наблюдении. Установлено наследование БГШ по аутосомно-рецессивному типу, что позволяет объяснить отсутствие аналогичной патологии в семье пациента у ближайших родственников.
Выделено два основных патоморфологических маркера БГШ — интенсивная коричневая пигментация и наличие сфероидных нейроаксональных образований в базальных ганглиях, преобладающих в области медиального сегмента бледного шара и ретикулярной части черной субстанции [14]. Формирование специфичной коричневой пигментации связано с локальным высоким содержанием железа и железосодержащего пигмента в межклеточном пространстве. Аксональные сфероиды при БГШ обнаруживаются не только в базальных ганглиях, но также в коре, белом веществе больших полушарий, спинном мозге, периферических нервах. Сфероиды представляют собой локальные расширения пресинаптических терминалей аксонов, которые содержат белки нейрофиламента, гранулы липофусцина, убиквитин, супероксиддисмутазу, ферритин, окисленное железо [6]. Эти патоморфологические находки позволяют отнести БГШ к группе болезней, названных синуклеинопатиями, к которым также относятся болезнь Паркинсона, деменция с тельцами Леви, мультисистемная атрофия [5].
Клинические проявления БГШ отличаются полиморфизмом симптомов (в том числе внутрисемейным), которые зависят от возраста начала заболевания и типа его течения [3, 11]. Обычно выделяют три клинические формы БГШ: 1) раннюю детскую (классическую) с дебютом в 4–10 лет; 2) ювенильную с началом в 10–18 лет; 3) взрослую (атипичную), развивающуюся после 18 лет [9]. Детская форма чаще проявляется после 5 лет дистонией нижних конечностей (в 90 %), затрудняющей ходьбу. В дальнейшем двигательные расстройства обычно генерализуются, вовлекая мышцы лица, глотки, туловища, формируя различные виды фокальной, мультифокальной, сегментарной и генерализованной дистонии. Cреди них могут встречаться блефароспазм, спастическая кривошея, синдром Мейжа, писчий спазм и их сочетания [11]. В 30 % случаев развивается синдром паркинсонизма в виде гипокинезии, ригидности и тремора покоя [10]. У части больных отмечаются зрительные расстройства в виде пигментной дегенерации сетчатки (29 %) и атрофии зрительных нервов (68 % больных) [9]. Характерны умственная отсталость вследствие ухудшения памяти, внимания и психические нарушения (агрессивность, асоциальное поведение). Эта форма отличается наиболее быстрым темпом прогрессирования с утратой способности к самостоятельному передвижению в течение 10–15 лет [13].
Ювенильная форма характеризуется дебютом в виде фокальной (конечностной, оромандибулярной) дистонии. У этих больных типичны нейропсихологические, поведенческие и когнитивные расстройства. Тип прогрессирования симптомов этой формы более медленный [2, 3].
Взрослая форма БГШ встречается реже (не более 15 % всех случаев болезни). В равной степени она проявляется различными видами гиперкинезов (дистония, хореоатетоз, гемибализм, миоклонии) и синдромом паркинсонизма [2]. К настоящему времени установлено, что для диагностики синдрома паркинсонизма необходимо, чтобы у больного отмечалась гипокинезия (облигатный признак), которая должна сопровождаться как минимум одним из следующих трех симптомов: повышением мышечного тонуса по типу «зубчатого колеса», тремором покоя и постуральной неустойчивостью (невозможностью удержать равновесие). Для оценки постуральной неустойчивости врач обычно становится позади больного и за плечи подталкивает его вперед, на себя и в стороны (проба Тавенарда). В норме пациент быстро восстанавливает равновесие, рефлекторно изменяя положение стоп, наклон туловища или совершает один-два корригирующих шага. При выраженном синдроме паркинсонизма больной с трудом удерживает равновесие, делает несколько мелких неэффективных шажков вперед (пропульсия), назад (ретропульсия), в сторону (латеропульсия) или падает без всякой попытки удержать равновесие.
В представленном наблюдении синдром паркинсонизма являлся ведущим в клинической картине и отличался рядом особенностей. Основным его проявлением была резко выраженная постуральная неустойчивость, формирование которой объясняют нарушением связей между бледным шаром и другими анатомическими образованиями (лобными долями, мозжечком, стволом) [6]. При этом описываемые большинством авторов замедленность движений с застываниями и экстрапирамидная ригидность в нашем наблюдении были выражены слабо, а различные виды гиперкинезов отсутствовали на протяжении всей болезни. Второй существенной особенностью экстрапирамидного синдрома были его симметричность и резистентность к препаратам леводопы, что также отмечено в подавляющем большинстве описываемых в литературе случаев БГШ [8, 10, 11]. Причиной последнего называют сохранность нигростриарного дофаминергического пути при этой патологии, что доказано результатами функциональной нейровизуализации [2].
Пирамидные симптомы, отмеченные в нашем наблюдении, являются довольно типичным признаком БГШ, хотя их выраженность обычно бывает гораздо меньше, чем при детской форме [9]. Сведения о наличии синдрома когнитивного дефицита у пациентов с БГШ противоречивы. С одной стороны, психические и поведенческие нарушения при взрослой форме описываются исследователями довольно часто, особенно депрессия, агрессивность, эмоциональная лабильность [2, 4, 6]. Их причины связывают с нарушением фронтостриарных путей, проходящих через бледный шар, которые участвуют в познавательной деятельности. Вместе с тем у части больных когнитивные функции остаются довольно сохранными, что имело место и в представленном случае. В редких случаях БГШ наблюдают парциальные либо вторично-генерализованные судорожные приступы [4]. У большинства пациентов с взрослой формой БГШ отмечают медленное течение болезни с сохранением функциональной активности в течение 15–40 лет [13, 15]. Однако в нашем наблюдении темп прогрессирования был необычно быстрым. Таким образом, представленный нами случай не укладывается в рамки разделения болезни на классическую и атипичную форму. В литературе классическая форма БГШ характеризуется ранним началом, быстрым прогрессированием, типичной клинической картиной, наличием изменений на МРТ в виде «глаза тигра». Для атипичной формы характерны позднее начало, медленное прогрессирование, более вариабельная клиническая картина и МРТ-признаки в виде «глаза тигра» либо гипоинтенсивной зоны в области бледного шара [15].
Очевидный полиморфизм неврологической симптоматики побудил исследователей к поиску унифицированных критериев диагностики БГШ. В результате предложены основные и дополнительные критерии для постановки диагноза БГШ. К основным признакам отнесены начало в первые два десятилетия жизни, прогрессирующее течение, наличие экстрапирамидных расстройств (дистония, ригидность или хореоатетоз), снижение интенсивности сигнала на МРТ от базальных ганглиев (особенно бледного шара и черной субстанции). Дополнительными симптомами заболевания являются пирамидные знаки (спастичность и патологические стопные рефлексы), прогрессирующее снижение интеллекта, пигментный ретинит, атрофия зрительных нервов, эпилептические припадки, положительный семейный анамнез, указывающий на аутосомно-рецессивный тип наследования [9]. Критериями исключения БГШ служат признаки иных болезней, способных объяснить данную клиническую картину: гепатоцеребральной дегенерации, болезни Гентингтона, других аутосомно-доминантных заболеваний. Несмотря на явный прогресс в определении дефиниций БГШ, некоторые из названных критериев не применимы к части случаев, в частности, возраст начала заболевания. Это связано с тем, что в литературе встречаются описания случаев начала БГШ у пациентов даже старше 30 лет, диагноз которых подтвержден с помощью ДНК-диагностики [14].
Существенную роль в диагностике БГШ отводят результатам нейровизуализации, в частности МР-томограммам головного мозга, получаемым с помощью высокопольных аппаратов с плотностью магнитного поля не менее 1–1,5 Тл. На Т2-взвешенных изображениях при этой патологии в 100 % типичных вариантов БГШ выявляют овальную симметричную гиперинтенсивную зону в переднемедиальной части внутреннего сегмента бледного шара («зрачок») внутри более обширной гипоинтенсивной зоны, которая названа «глазом тигра» [8]. Повышение интенсивности сигнала в литературе объясняют глиозом и уменьшением количества клеток с низким содержанием железа, напротив, снижение интенсивности сигнала — увеличением концентрации железа более 2 мг [12]. Сроки появления данного типичного феномена при БГШ дискутируются. По мнению ряда авторов, этот симптом может опережать клинические проявления заболевания либо появляться спустя 2–3 года от начала болезни [10]. Применение позитронно-эмиссионной томографии позволяет у больных с БГШ обнаружить регионарное снижение церебрального кровотока в лобно-теменных долях, бледном шаре и стиатуме [4].
К настоящему времени эффективного лечения БГШ не существует. С учетом известного патогенеза предпринята попытка применения хелатных соединений, однако она не увенчалась успехом, так как при БГШ наблюдается лишь локальное увеличение содержания железа в головном мозге, в то же время его обмен в целом не страдает. Использование антиоксидантов с целью возможного их влияния на антиоксидантный стресс также не привело к положительным результатам. В связи с этим лечение данной патологии носит только симптоматический характер [2]. При синдроме паркинсонизма можно назначать в индивидуальных дозах дофаминовые агонисты (мирапекс, проноран) или амантадины (мидантан, ПК-Мерц). В случаях гиперкинезов рекомендуют атипичные бензодиазепины (клоназепам), вальпроаты (депакин, энкорат) или инъекции диспорта в заинтересованные мышцы. При спастичности применяют миорелаксанты (мидокалм, баклофен). С целью коррекции когнитивных нарушений назначают средства, влияющие на нейромедиаторный обмен (нейромидин, глиатилин). Для коррекции психотических нарушений могут использоваться атипичные нейролептики (клозапин) или селективные ингибиторы обратного захвата серотонина (венлафаксин). В случаях эпилептического синдрома показаны вальпроаты или топамакс. Перспективным направлением терапии БГШ является использование пантотеновой кислоты (витамин В5) и метода глубокой магнитной стимуляции бледного шара [10, 13].
1. Болезни нервной системы. Руководство для врачей / Под ред. Н.Н. Яхно. — М.: Медицина, 2005. — Т. 2. — С. 170-171.
2. Левин О.С., Юнищенко Н.А., Амосова Н.А. и др. Болезнь Галлервордена — Шпатца с поздним началом // Неврол. журн. — 2004. — № 2. — С. 36-46.
3. Неординарные (раритетные) синдромы и заболевания нервной системы у детей и взрослых / Под ред. проф. С.К. Евтушенко. — Донецк — Святогорск, 2003. — 285 с.
4. Юдина Г.К., Шоломов И.И. Семейный случай болезни Галлервордена — Шпатца // Журн неврол. и психиатр. — 2003. — № 1. — С. 49-50.
5. Doi H., Koyano S., Miyatake S. et al. Siblings with the adult-onset slowly progressive type of PKAN and a novel mutation: clinical features and (99m) Tc-ECD brain perfusion SPECT findings // J. Neurol. Sci. — 2010. — № 1–2. — P. 172-176.
6. Galvin J.E. Neurodegenerative Diseases: Pathology and the Advantage of single-Cell Profiling // Neurochemical Research. — 2004. — Vol. 29, № 6. — P. 1041-1051.
7. Gregory A., Polster B., Hayflick S. et al. Clinical and genetic delineation of neurodegeneration with brain iron accumulation // J. Med. Genet. — 2009. — № 2. — P. 73-80.
8. Gupta R., Kumar A., Sharma M. et al. Autopsy always teach and tell: neurodegeneration with brain iron accumulation: a case report // Indian J. Pathol. Microbiol. — 2007. — № 4. — P. 792-794.
9. McNeill A., Birchall D., Hayflick S. et al. MRI distinguishes foir subtypes of neurodegeneration with brain iron accumulation // Neurology. — 2008. — № 18. — P. 1614-1619.
10. Kapoor S., Hortnagel K., Gogia S. et al. Pantothenate Kinase Associated Neurodegeneration (Hallervorden — Spatz Syndrome) // Indian J. Pediatr. — 2005. — № 3. — P. 261-263.
11. Sachin S., Goyal V., Singh S. et al. Clinical spectrum of Hallervorden — Spatz Syndrome in India // J. Clin. Neurosci. — 2009. — № 2. — P. 253-258.
12. Szumowski J., Bas E., Gaarder K. et al. Measurement of brain iron distribution in Hallervorden — Spatz Syndrome // J. Magn. Reson. Imaging. — 2010. — № 2. — P. 482-489.
13. Textbook of Clinical Neuropsychiatry / Еd. D.P. Moor. — UK: Hodder Arnos, 2008. — P. 364-365.
14. Vincze A., Kapas I., Molnar M. et al. Clinicopathological variability in neurodegeneration with brain iron accumulation // Ideggyogy Sz. — 2010. — № 3–4. — P. 129-135.
15. Zolkipli Z., Dahmoush H., Saunders D. et al. Pantothenate kinase 2 mutation with classic PKAN without «eye-of-the-tiger» sign on MRI in pair of siblings // Pediatr. Radiol. — 2006. — № 36. — P. 884-886.