
Международный неврологический журнал 5 (43) 2011
Вернуться к номеру
Симпозиум «Болезнь Паркинсона (клиника, диагностика, принципы терапии)»
Авторы: Луцкий И.С., Евтушенко С.К., Симонян В.А., Донецкий национальный медицинский университет им. М. Горького
Рубрики: Неврология
Версия для печати
Проводит: кафедра детской и общей неврологии ФИПО Донецкого национального медицинского университета им. М. Горького.
Рекомендован: неврологам, семейным врачам, терапевтам.
Экстрапирамидная система остается одной из наименее изученных систем головного мозга с позиций как фундаментальной, так и клинической неврологии. Указанная ситуация сложилась вследствие ряда объективных факторов: сложности анатомического строения, многочисленных нейрональных связей с различными отделами нервной системы и наличия значительного числа интернейронов, многоуровневой системы функционирования, в которой задействовано значительное количество различных нейротрансмиттеров, трудностей лабораторной и инструментальной объективизации клинических синдромов.
Экстрапирамидная система представляет собой анатомо-функциональную систему, которая включает базальные ганглии (по определению некоторых исследователей, базальные ганглии относят к высшим центрам экстрапирамидной системы), часть серого вещества среднего и промежуточного мозга, многочисленные связи этих структур с различными отделами головного и спинного мозга. Впервые термин «экстрапирамидная система» предложил Wilson (1912) после описания им гепатолентикулярной дегенерации.
К базальным ганглиям относится группа ядер, расположенных в основании полушарий большого, среднего и промежуточного мозга (рис. 1):
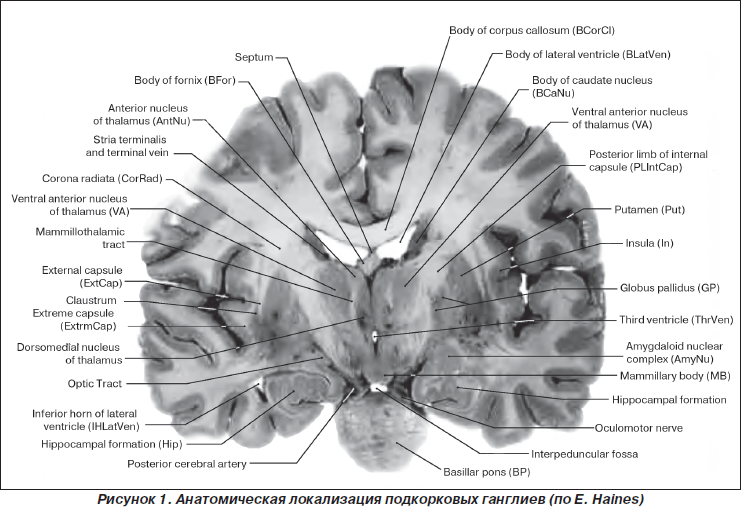
I. Хвостатое ядро (nucleus caudatus).
II. Чечевицеобразное ядро (nucleus lentiformis), состоящее:
— из скорлупы (putamen);
— бледного шара (globus pallidum).
III. Черная субстанция (substancia nigra).
IV. Субталамическое ядро (nucleus subthalamicus).
Детальное описание базальных ганглиев принадлежит В.М. Бехтереву, который объединил хвостатое ядро и скорлупу в неостриатум, или стриатум (филогенетически более молодые образования), бледный шар определил как палеостриатум, или палидум. В настоящее время установлено, что функцию стриатума во многом определяет его клеточный состав. Подсчитано, что в стриатуме человека содержится около 110 миллионов мелких ахроматических нейронов и около 670 тысяч крупных мультиполярных нейронов. 95 % клеток стриатума представлены шиповидными нейронами среднего размера, которые являются проекционными нейронами. Остальные нейроны относятся к интернейронам (вставочным).
Весь поток афферентной информации, поступающей в стриопаллидарный комплекс, оканчивается в стриатуме. Он получает импульсацию из следующих структур:
1) большинства областей коры большого мозга;
2) центральных интраламинарных ядер таламуса;
3) компактная часть черной субстанции и других ядер вентральной области покрышки среднего мозга;
4) ядер шва среднего мозга;
5) голубого пятна.
Из стриатума информация поступает к бледному шару и ретикулярной части черной субстанции, от которых начинаются основные эфферентные пути базальных ганглиев к двигательным областям таламуса, ядрам ретикулярной формации среднего мозга, главным образом к педункулопонтинному ядру, являющемуся передаточным звеном влияния базальных ганглиев на спинальные двигательные механизмы.
Огромное значение в понимании функционирования базальных ганглиев, их роли в регуляции двигательных функций и развитии двигательных нарушений связано с изучением их нейротрансмиттерной организации. Установлено, что функциональная активность стриопаллидарной системы определяется сложным взаимодействием различных нейротрансмиттерных систем. В настоящее время наиболее полно изучена функция следующих нейротрансмиттеров: глутамата, аспартата, гамма-аминомасляной кислоты, норадреналина, серотонина, адреналина, дофамина, субстанции Р.
Обмен информацией между корой и базальными ганглиями осуществляется посредством корково-подкорковых кругов. В настоящее время известно пять таких кругов, из них два участвуют в регуляции движений (сенсомоторный и окуломоторный), а три — в регуляции когнитивных функций и поведенческих реакций (дорсолатеральный префронтальный, латеральный орбитофронтальный, передний поясной). При болезни Паркинсона патология указанных немоторных путей играет роль в формировании психических нарушений.
Основную роль в формировании двигательных нарушений при патологии экстрапирамидной системы играет патология нигростриарного дофаминергического пути. Дегенерация этого пути при болезни Паркинсона приводит к резкому снижению синтеза и высвобождения дофамина из его терминалей в стриатуме. Функционирование нигростриарного дофаминергического пути зависит от активности дофаминовых рецепторов. В настоящее время выделяют две их большие группы: D1- и D2-рецепторы.
Существуют еще две восходящие дофаминергические системы: мезолимбический и мезокортикальный пути. Мезолимбический путь связывает средний мозг с филогенетически более старыми образованиями переднего мозга и оканчивается в вентральной области стриатума и лобной коре. Считается, что основной функцией этого пути является участие в контроле настроения и поведенческих реакций. Дефицит дофамина в мезолимбическом пути наблюдается при болезни Паркинсона, что может объяснить часто встречающиеся при этом заболевании эмоциональные (прежде всего депрессия) и другие психические нарушения. Кроме того, имеются сведения об участии мезолимбического пути в контроле начала двигательного акта и двигательных аффективных реакций. Следовательно, его дисфункция может иметь значение в развитии первичной акинезии.
Мезокортикальный дофаминергический путь идет от среднего мозга к префронтальной, поясной и обонятельной областям коры больших полушарий. Предполагается, что его активация может быть связана с тормозящим воздействием на поведенческую активность. Считают, что патология этого пути совместно с патологией мезокортикального пути может иметь значение в формировании психических нарушений при болезни Паркинсона.
Следует подробнее остановиться на связи стриатума с бледным шаром. Она осуществляется посредством двух нейрональных путей. Один из них прямой, он связывает скорлупу и хвостатое ядро с внутренним сегментом бледного шара и ретикулярной частью черной субстанции. Его функция преимущественно регулируется D1-рецепторами. Другой путь непрямой: он достигает внутреннего сегмента бледного шара и ретикулярной части черной субстанции, направляясь вначале к наружному сегменту бледного шара, а затем к субталамическому ядру, и лишь после этого достигает своей первоначальной цели. Непрямой путь регулируется преимущественно D2-рецепторами.
Активация прямого пути стимулирует моторные отделы коры и облегчает формирование движений, а непрямого — ослабляет возбуждающие таламокортикальные влияния и вызывает гипокинезию и ригидность. Дофамин облегчает проведение импульсов по прямому пути и оказывает ингибирующее воздействие на непрямой путь. В условиях дефицита дофамина (болезнь Паркинсона) происходит снижение функциональной активности прямого пути и повышение активности непрямого пути. Это сопровождается торможением проведения по таламокортикальному возбуждающему пути и нарушением функциональной активности дополнительной моторной коры.
Болезнь Паркинсона (БП) представляет собой хроническое прогрессирующее заболевание головного мозга с дегенерацией нигростриарных нейронов и нарушением функции базальных ганглиев. Впервые заболевание описал английский невролог Джеймс Паркинсон в 1817 году и назвал его «дрожательным параличом». В 1877 году невролог Шарко более подробно описал клинические проявления заболевания и предложил называть его болезнью Паркинсона. БП — одна из самых частых форм первичных хронических нейродегенеративных заболеваний. По современным представлениям БП, или первичный идиопатический паркинсонизм, является как спорадическим, так и семейным нейродегенеративным мультисистемным расстройством со значительным клиническим полиморфизмом и вариативностью течения, этиопатогенетической и морфологической неоднородностью, с хорошим эффектом дофаминергической терапии по отношению к классическим паркинсоническим двигательным нарушениям.
Распространенность БП достаточно высока и колеблется от 67 до 350 случаев на 100 тыс. населения. Самая высокая распространенность зарегистрирована в США —107–329 случаев на 100 тыс. населения, самая низкая из европейских стран — в Швеции — 76 случаев. В Украине распространенность составляет около 133 случаев на 100 тыс. населения, хотя реальные цифры представляются значительно более высокими. Заболевание имеет четкую возрастзависимую структуру: чем старше возрастная популяция населения, тем чаще встречается заболевание. Так, в возрасте после 65 лет страдает 1 % возрастной популяции, в возрасте после 80 лет заболевание встречается у 3–4 % населения. Наиболее часто первые симптомы заболевания регистрируются в возрасте 42–52 лет. Следует подчеркнуть, что, несмотря на традиционные представления о возрастзависимом характере болезни Паркинсона, случаи заболеваемости в более молодом возрасте уже давно не являются редкостью: считается, что примерно каждый десятый пациент заболевает болезнью Паркинсона в возрасте до 50 лет, а каждый двадцатый — до 40 лет. В связи с этим выделяют даже отдельную подгруппу — болезнь Паркинсона с ранним началом, отличающуюся рядом особенностей механизмов развития болезни, а также клинической картиной и течением, реакцией на противопаркинсонические препараты, прогнозом. В настоящее время в мире насчитывается около 5–6 миллионов больных БП. Заболеваемость на 100 тыс. населения также имеет различную тенденцию в зависимости от страны, где проводилось исследование. Самая высокая заболеваемость зарегистрирована на Фарерских островах — 21,1, самая низкая — в Ливии — 4,5 на 100 тыс. населения. Мужчины и женщины болеют приблизительно с одинаковой частотой, с некоторым преобладанием мужчин, хотя в Японии женщины болеют в 1,5 раза чаще.
Только в начале XXI века прогресс в развитии молекулярной генетики и молекулярной биологии позволил пролить свет на генетические основы этиологии БП. Была проведена идентификация ряда генов наследственных форм первичного паркинсонизма в семейных случаях. По современным представлениям, от 5 до 10 % всех случаев БП имеют прямую моногенную основу. Остальные случаи представлены спорадической формой и имеют мультифокальную природу. В развитии спорадической формы БП играет решающую роль взаимодействие генетических и средовых факторов, что в конечном итоге определяет особенности клеточной детоксикации и обмена ксенобиотиков, антиоксидантной защиты, процессинга ряда нейрональных белков, характера дофаминового обмена. Раскрытие основных молекулярных звеньев нигростриарной дегенерации позволило прийти к пониманию патобиохимического каскада при БП.
В настоящее время идентифицировано более 15 генов наследственных форм первичного паркинсонизма. Наиболее изученными и имеющими ведущее значение являются 6 генетических вариантов:
1. PARK1 (a-синуклеин). Белок альфа-синуклеин играет важную роль в синаптическом везикулярном транспорте и хранении нейротрансмиттеров. Мутации (наследственные или вследствие воздействия экзогенных нейротоксических факторов) в гене a-синуклеина приводят к изменению структуры белка, его накоплению в нейроне и агрегации с образованием телец Леви. В настоящее время a-синуклеин рассматривается в качестве ключевого молекулярного маркера патологии нейронов и модуляции процессов нейродегенерации паркинсонического типа.
2. PARK2 (паркин). Мутации в гене паркина являются частой причиной раннего, в том числе ювенильного, паркинсонизма (до 50 % семейных форм и около 15 % спорадических случаев). Паркин представляет собой убиквитин-протеинлигазу типа Е3, функция которой заключается в доставке аномально конформированных белков в протеасомный комплекс для последующего расщепления.
3. PARK6 (PINKI). Форма аутосомно-рецессивного паркинсонизма обеспечивает развитие до 9 % случаев раннего начала заболевания. Белок PINKI является митохондриальной протеинкиназой и играет важную роль в митохондриальном биогенезе.
4. PARK7 (DJ-I). Редкая форма аутосомно-рецессивного паркинсонизма — 1–2 % ранних случаев БП. Белок DJ-I играет важную роль в поддержании целостности и выживаемости дофаминергических нейронов.
5. PARK8 (LRRK2). Ген связан с аутосомно-доминантной формой паркинсонизма с пенетрантностью до 40 %. Ген LRRK2 имеет большое значение в развитии спорадических случаев первичного паркинсонизма: от типичной поздней БП с тельцами Леви до атипичных вариантов синуклеин- и тау-патологии. Белковым продуктом гена является дардарин, именно его патологическая активация является следствием доминантной мутации в гене LRRK2 и приводит к развитию нейродегенеративных изменений.
6. Ген GBA. Кодирует лизосомальный фермент глюкоцереброзидазу. Мутации в указанном гене могут сопровождаться развитием различных вариантов синуклеинопатий — классической БП и деменции с тельцами Леви.
Во многом вопросы патогенеза БП остаются неизвестными, и их предстоит еще изучить. Следует упомянуть об этапных открытиях, которые меняли наши представления о природе БП. Среди них — описание дегенерации крупных мелатонинсодержащих клеток черной субстанции, которое принадлежит русскому ученому К.П. Третьякову (1919), выявление дефицита дофамина в базальных ганглиях (H. Ehringer, O. Hornykiewicz, 1960). Обобщив материалы различных авторов по изучению дегенерации нигростриарных нейронов, Г.Н. Крыжановский и соавт. (1995) перечислили расстройства внутриклеточного метаболизма, способствующие возникновению нейродегенерации:
— нарушение митохондриального дыхания и повреждение митохондрий;
— энергетический дефицит нейрона;
— усиление свободнорадикального окисления с образованием агрессивных перекисей;
— избыточное накопление свободных ионов кальция;
— нарушение метаболизма ксенобиотиков.
Важнейшим этапом в понимании патогенеза БП явились колоссальные успехи, достигнутые в изучении генетических факторов развития болезни, приводящих к развитию нейродегенеративного процесса (рис. 2). Установлено, что в основе молекулярных механизмов БП лежит нарушение системы контроля за биогенезом, пространственной организацией и биодегенерацией нейрональных белков. Ключевая роль отводится a-синуклеину, патологическая агрегация которого в виде фибриллярных структур является первым этапом формирования телец Леви. При этом наибольшей нейротоксичностью обладают промежуточные олигомерные формы a-синуклеина, тогда как формирование зрелых фибрилл, и тем более телец Леви, является защитной реакцией клетки, направленной на нейтрализацию токсичных a-синуклеиновых олигомеров. Нейротоксичность реализуется за счет различных механизмов: индукции свободнорадикальных реакций, нарушений эндоплазматического тока, активации стрессовых протеинкиназ и процессов апоптоза, активации микроглии, нарушения взаимодействия альфа-синуклеина с его естественными белками-партнерами. Конечным результатом этих процессов является развитие и прогрессирование нейродегенеративных процессов с уменьшением продукции дофамина. Как указывалось выше, это вызывает дисфункцию нейронов базальных ганглиев, прежде всего растормаживание и избыточную активность нейронов внутреннего сегмента бледного шара, ретикулярной части черной субстанции, и приводит к торможению таламокортикальных нейронов и дефициту активации нейронов дополнительной моторной коры, с которыми связывают развитие основных моторных проявлений БП. Помимо дофаминергических нейронов черной субстанции, при БП дегенерации подвергаются и другие группы нейронов, в том числе нейроны дорсального ядра блуждающего нерва, нейроны обонятельной луковицы, норадренергические нейроны голубого пятна, серотонинергические нейроны ядер шва, холинергические нейроны ядра Мейнерта, а также нейроны коры больших полушарий и некоторые вегетативные сплетения. В силу этого, помимо дефицита дофамина, возникает дисфункция серотонинергических, норадренергических и холинергических систем. С поражением экстранигральных структур связаны немоторные проявления болезни.
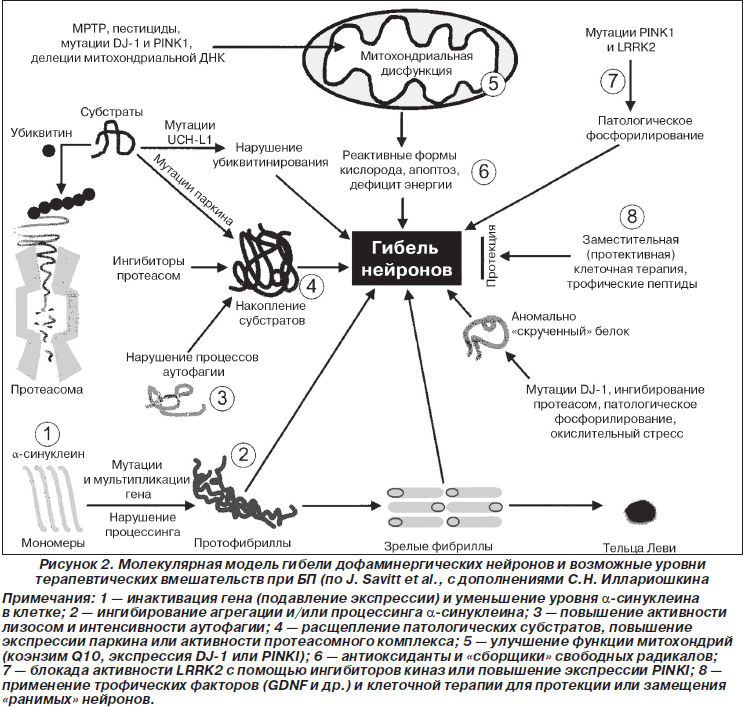
Клинические проявления БП
Основу клинической картины БП составляет классическая триада: акинезия (гипокинезия), мышечная ригидность и тремор покоя. На более поздней стадии заболевания присоединяется четвертый симптом — постуральная неустойчивость. Заболевание может начаться с каждого из трех указанных симптомов, к которому впоследствии присоединяются остальные симптомы. В некоторых случаях дебют БП может состоять сразу из двух или даже трех симптомов.
Акинезия (гипокинезия) представляет собой снижение спонтанной двигательной активности. Истинная акинезия (полное отсутствие движений) встречается достаточно редко, преимущественно на заключительных стадиях БП. На начальных этапах наблюдается замедленность (брадикинезия) и бедность произвольных движений (олигокинезия).
Выделяют четыре компонента акинезии (A. Barbeau, 1984):
1) нарушение моторной инициативы;
2) нарушение кинетической «мелодии»;
3) нарушение стратегии двигательного обучения;
4) быстрая истощаемость при выполнении повторных заданий с постепенным затуханием двигательной активности.
Клинически гипокинезия выявляется при целом ряде произвольных движений:
— больной застывает в различных позах, иногда напоминая манекен;
— возникают затруднения при инициации движения и перемене положения тела (если гипокинезия достаточно выражена, больной может делать несколько попыток, прежде чем совершить движение);
— развивается гипомимия, отсутствует эмоциональная окраска на лице, больной редко моргает (симптом Мари);
— речь становится тихой, монотонной, маломодулированной, постепенно затухающей, из-за тремора голосовых связок речь может приобретать «тремолирующий» характер;
— характерно развитие нарушений фонации (дисфония), в выраженных случаях речь становится шепотной. Некоторые пациенты по наличию или отсутствию нарушений фонации самостоятельно регулируют дозу левадопосодержащих препаратов;
— во время ходьбы отсутствуют содружественные движения (ахейрокинез), при гемипаркинсонизме — на одной стороне.
По механизму развития некоторые исследователи выделяют первичную и вторичную акинезию. Вторичная акинезия вызвана ригидностью и связана с первичной дегенерацией нейронов вентрального отдела компактной части черной субстанции. Первичная акинезия развивается позже, в среднем через 5 лет после начала болезни, и связана с гибелью нейронов дорсального слоя и медиального ядра компактной части черной субстанции (дофаминовое обеспечение мезолимбического дофаминергического пути). Она сопровождается психомоторной акинезией, депрессией и формированием когнитивных нарушений. С развитием первичной акинезии снижается эффективность препаратов леводопы.
Для раннего выявления гипокинезии возможно применение следующих тестов:
— тест Фурнье: пациенту предлагают максимально быстро совершать серию движений: встать, сесть, повернуться, наклониться и т.п. Уже на ранних стадиях гипокинезии при выполнении теста можно заметить замедленность движений;
— тест постукивания большим и указательным пальцами: пациент в максимально возможном темпе и с максимальной амплитудой выполняет постукивания по столу большим и указательным пальцами поочередно обеими руками. Тест особенно информативен при формировании гемипаркинсонизма — при этом отстает от темпа и амплитуды одна рука;
— тест сжимания и разжимания кисти: пациенту предлагается максимально быстро сжимать и разжимать кисть (отстает кисть на стороне формирующейся гипокинезии).
Следующий симптом — ригидность (мышечная гипертония) — представляет собой пластическое повышение мышечного тонуса и имеет следующие особенности:
— характеризуется повышением мышечного тонуса уже в начальной фазе движения и сохраняется до конца движения, обусловлена одновременным сокращением мышц-антагонистов и агонистов. Характерно нарастание степени ригидности от движения к движению. Описанная мышечная ригидность определяется как пластический мышечный гипертонус, или экстрапирамидная ригидность;
— феномен Негро — симптом «зубчатого колеса»: ощущение прерывистости, ступенчатости сопротивления мышц на фоне пластического гипертонуса;
— симптом «воздушной подушки»: после поднятия головы больного над подушкой она остается некоторое время в этом положении;
— поза «просителя»: из-за повышенного мышечного тонуса голова опущена, руки и ноги подогнуты в локтевых и коленных суставах;
— симптом Дылева: сила пассивного сопротивления значительно больше силы активных движений;
— при выполнении теста маятникообразного качания ног — прекращение качания сразу после выполнения теста.
Для выявления ригидности в начальных стадиях заболевания, когда она выражена незначительно, можно использовать следующие тесты:
— прием Нойка — Ганева: при выполнении врачом пассивных сгибательно-разгибательных движений в лучезапястном суставе у лежащего больного его просят медленно поднимать ногу на стороне исследования — при наличии скрытого пластического тонуса исследователь ощущает увеличение сопротивления в исследуемой конечности;
— симптом Форманна: нарастание пластически повышенного мышечного тонуса при его исследовании в руках у пациента в позе Ромберга с закрытыми глазами;
— пластическое повышение мышечного тонуса нарастает после повторных движений в исследуемой конечности;
— тест «встряхивания за плечи»: пациент принимает вертикальное положение, его просят расслабиться, затем врач кладет ладони на плечи больного и совершает быстрые попеременные вращательные полуобороты туловища его вокруг вертикальной оси. Тест является достаточно чувствительным для оценки степени мышечной ригидности и гипотонии, позволяет определить симметричность мышечно-тонических изменений.
Тремор (дрожание). По своему характеру определяется как тремор покоя:
— частота тремора составляет 4–6 Гц;
— обычно начинается с одной стороны с дистальных отделов руки;
— в кисти разнонаправленные движения большого пальца и остальных пальцев кисти создают своеобразную картину «скатывания пилюль» или «счета монет»;
— в типичных случаях тремор покоя пропадает во время произвольных движений и исчезает во сне;
— при прогрессировании болезни дрожание последовательно распространяется с кисти на предплечье, плечо (лучезапястный, локтевой, плечевой суставы), в последующем в этот процесс может вовлекаться одноименная нога. Дрожание в ноге можно наблюдать, когда больной сидит в неудобной позе, во время ходьбы тремор исчезает;
— в некоторых случаях при генерализации тремора в дрожание вовлекаются мышцы шеи и лица, что сопровождается дрожанием головы, нижней челюсти, губ, языка.
Постуральные нарушения. Складываются из нарушений позы, статики и походки. Они обусловлены гипокинезией, мышечной ригидностью, дисфункцией постурального тонуса и рефлексов; клинически проявляются в следующем:
— больной с трудом удерживает центр тяжести тела в площади опоры;
— возникает феномен пропульсии, ретропульсии, латеропульсии: внезапный спонтанный или вызванный внешним толчком переход к быстрой ходьбе вперед, назад или в сторону. При этом тело пациента наклонено в сторону движения и может опережать движение ног, что сопровождается падением больного;
— симптом «топтания»: больной, встав со стула или с кровати, не может сразу начать движение, а некоторое время топчется на одном месте. Начав движение, он идет мелкими шажками (микробазия), шаркая ногами по полу. Для того чтобы изменить направление движения, больной вынужден остановиться, некоторое время топчется на месте и лишь постепенно меняет направление и начинает движение. При поворотах возможно падение больного.
Парадоксальные кинезии. Иногда на фоне гипокинезии возникают эпизоды, когда больной может внезапно совершать ряд быстрых сложных движений (побежать за троллейбусом, танцевать). Обычно это происходит в состоянии аффекта либо на фоне эмоциональных переживаний, чаще на фоне положительных эмоций. Период парадоксальной кинезии затем вновь сменяется акинетико-ригидным синдромом, который на некоторое время принимает более выраженный характер.
В настоящее время считается общепризнанным, что БП проявляется клинически не только двигательными нарушениями, но и имеет целый спектр недвигательных проявлений, которые встречаются у всех пациентов независимо от возраста дебюта заболевания и стадии болезни:
1) нервно-психические нарушения:
— эмоциональные;
— когнитивные;
— психотические;
— поведенческие;
2) нарушения сна и бодрствования;
3) вегетативные нарушения;
4) сенсорные нарушения и боль;
5) повышенная утомляемость.
Большинство немоторных проявлений появляются и нарастают по мере прогрессирования заболевания — параллельно с усугублением двигательных расстройств. Но некоторые немоторные проявления, такие как нарушение обоняния, запоры, нарушения сна, болевые синдромы, возникают до развития классических моторных симптомов БП. В связи с этим в настоящее время говорят о доклинической, премоторной стадии БП.
Понимание механизмов развития и роли немоторных проявлений в структуре БП отражает концепция H. Braak и соавт. (2002), согласно которой дегенеративный процесс не ограничивается компактной частью черной субстанции, а последовательно вовлекает большое количество различных моторных структур головного мозга. H. Braak и соавт. выделили 6 стадий развития патологического процесса при БП (рис. 3). Первая стадия характеризуется дегенерацией обонятельной луковицы и переднего обонятельного ядра, которая клинически может проявляться нарушением обоняния. Вторая стадия характеризуется вовлечением ядер ствола мозга, контролирующих аффективные, вегетативные функции, цикл сна и бодрствования, и может проявляться расстройством поведения во сне с быстрыми движениями глаз, депрессией, запорами. Классические моторные проявления болезни появляются только на третьей и четвертой стадиях по H. Braak, что связано с распространением дегенеративного процесса на черную субстанцию. В финальных пятой и шестой стадиях тельца Леви появляются в лимбических структурах и коре головного мозга, что приводит к развитию когнитивных, поведенческих и психомоторных расстройств.
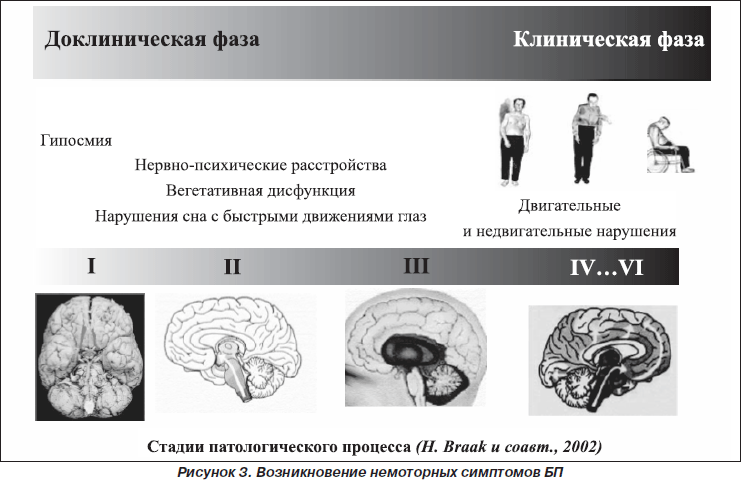
Значительная часть немоторных проявлений резистентна к препаратам леводопы, что указывает на их связь с дисфункцией недофаминергических систем: норадренергических, серотонинергических, холинергических и других.
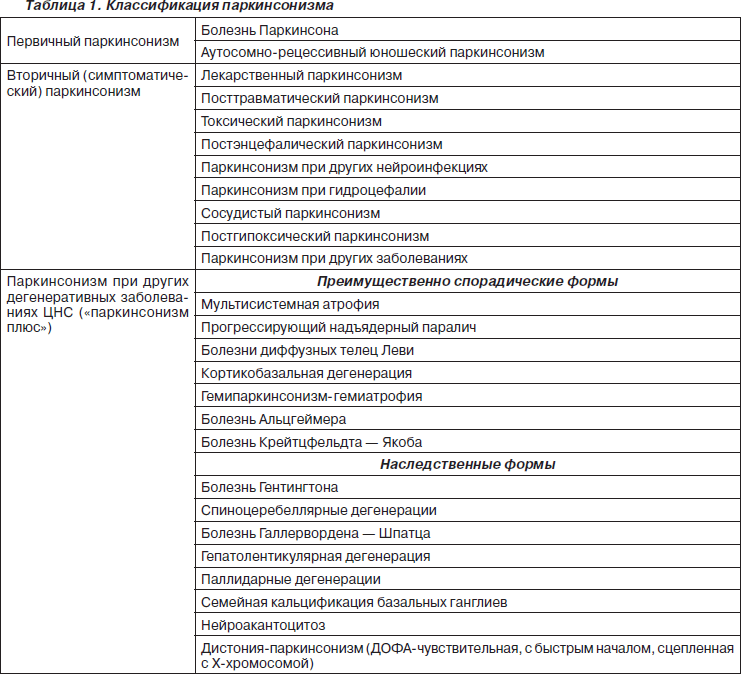
Классификация паркинсонизма
Классификация построена с учетом того, что паркинсонизм представляет собой полиэтиологический синдром. Выделяют идиопатический (первичный) паркинсонизм, в структуру которого входит БП и более редкий идиопатический паркинсонизм, имеющий генетическую основу, вторичный паркинсонизм — вследствие каких-либо поражений или заболеваний ЦНС (сосудистый, травматический, постэнцефалитический и др.). При «паркинсонизме плюс» паркинсонический синдром входит в структуру других нейродегенеративных заболеваний и ряда заболеваний нервной системы наследственного характера (табл. 1).
Клинические формы БП в классическом виде соответствуют трем основным симптомам: дрожательная, ригидная и акинетическая формы могут наблюдаться только в начальной стадии заболевания. По мере прогрессирования болезни в клинической картине наблюдается сочетание указанных симптомов. В зависимости от преобладания того или иного симптома выделяют следующие формы: смешанную (акинетико-ригидно-дрожательную), акинетико-ригидную и дрожательную. Смешанная форма выявляется в 60–70 % случаев болезни Паркинсона, акинетико-ригидная — в 15–20 %, дрожательная — в 5–10 % случаев. По мере прогрессирования заболевания его клиническая форма может меняться.
В течении БП выделяют стадии развитии. Общепризнанной является классификация, предложенная Hoehn и Yarh (1967):
— стадия 1. Односторонние проявления заболевания (только конечности);
— стадия 1.5. Односторонний процесс (конечности и одноименная сторона туловища);
— стадия 2. Двустороннее заболевание без постуральной неустойчивости;
— стадия 2.5. Начальные проявления двустороннего процесса с возвращением к норме при исследовании;
— стадия 3. Умеренно выраженная постуральная неустойчивость, возможно самостоятельное передвижение;
— стадия 4. Значительная утрата двигательной активности, пациент не в состоянии передвигаться без посторонней помощи;
— стадия 5. При отсутствии посторонней помощи пациент прикован к постели или инвалидному креслу.
Учитывая интенсивность развития симптомов БП, выделяют три варианта темпа прогрессирования болезни (при условии адекватного лечения):
— быстрый темп прогрессирования, при котором смена стадий заболевания (первая — вторая или вторая — третья) происходит в течение 2 или менее лет;
— умеренный темп прогрессирования, при котором смена стадий происходит более чем через 2 года, но не более чем в течение 5 лет;
— медленный темп прогрессирования со сменой стадий более чем через 5 лет.
Клиническая диагностика БП включает три этапа:
1. Распознавание экстрапирамидного синдрома.
2. Уточнение анамнестических данных, выявление сопутствующих синдромов, лабораторные исследования, нейровизуализация (СКТ, МРТ, ПЭТ), леводопа-диагностика, ЭНМГ-диагностика.
3. Установление нозологического диагноза (при паркинсонизме — определение степени тяжести по рейтинговым шкалам).
В настоящее время для клинической диагностики болезни Паркинсона используют критерии включения, предложенные банком головного мозга Общества болезни Паркинсона Великобритании (A. Hughes et al., 1992).
Диагностика синдрома паркинсонизма
Критерии включения болезни Паркинсона: гипокинезия в сочетании с не менее чем одним из следующих симптомов:
— мышечная ригидность;
— тремор покоя 4–6 Гц;
— постуральная неустойчивость, не связанная с первичными зрительными, вестибулярными, мозжечковыми или проприоцептивными нарушениями.
Критерии исключения болезни Паркинсона:
— повторные инсульты в анамнезе со ступенеобразным прогрессированием симптомов паркинсонизма;
— повторные черепно-мозговые травмы в анамнезе;
— энцефалит в анамнезе;
— окулогирные кризы;
— лечение нейролептиками на момент появления симптомов;
— семейный характер заболевания (более 1 родственника с аналогичным заболеванием);
— наличие длительной ремиссии;
— строго односторонняя симптоматика более 3 лет;
— паралич взора вниз;
— ранняя быстро прогрессирующая вегетативная недостаточность;
— мозжечковые знаки;
— рано развивающаяся деменция с нарушениями памяти, речи, праксиса;
— симптом Бабинского;
— наличие атрофии мозжечка или сообщающейся гидроцефалии при компьютерной томографии;
— отсутствие реакции на высокие дозы леводопы (при исключении мальабсорбции);
— контакт с токсическими веществами, вызывающими паркинсонизм.
Критерии, подтверждающие диагноз болезни Паркинсона (не менее трех):
— одностороннее начало;
— тремор покоя;
— прогрессирующее течение;
— сохранение асимметрии симптоматики с преобладанием на первоначально вовлеченной стороне;
— высокая эффективность препаратов леводопы (уменьшение симптомов на 70–100 %);
— выраженные хореиформные дискинезии, индуцированные леводопой;
— сохранение реакции на леводопу в течение 5 лет и более;
— продолжительность заболевания в течение 10 лет и более.
Лекарственный паркинсонизм является одним из самых частых форм вторичного паркинсонизма и составляет от 4 до 10 % всех случаев заболевания. Наиболее часто он связан с применением нейролептиков (нейролептический паркинсонизм), поэтому его распространенность наиболее высока среди пациентов психиатрических клиник и амбулаторных психиатрических учреждений.
Реже развитие лекарственного паркинсонизма вызывают другие лекарственные средства:
— другие агонисты дофаминовых рецепторов (метоклопрамид, дипразин, флунаризин, циннаризин);
— препараты, снижающие кругооборот дофамина в синапсах (метилдофа);
— центральные симпатолитики, истощающие запас дофамина в нервных терминалях (резерпин, тетрабеназин);
— серотонинергические средства, тормозящие активность дофаминергических нейронов черной субстанции.
Нейролептический паркинсонизм возникает у 10–15 % пациентов, принимающих нейролептики. Вероятность развития паркинсонизма зависит от способности препарата вызывать блокирование D2-рецепторов. Наиболее часто развитие побочного эффекта наблюдается при приеме неселективных нейролептиков (галоперидол, аминазин, трифтазин). По данным позитронно-эмиссионной томографии, симптомы паркинсонизма возникают при блокировании не менее 80 % D2-рецепторов. Наиболее часто возникают у лиц старше 40 лет, у женщин — в 2 раза чаще. Наличие органического поражения головного мозга увеличивает вероятность развития нейролептического паркинсонизма. Симптомы паркинсонизма развиваются через несколько дней или недель после начала приема препарата. Большие дозы нейролептиков, резкое увеличение дозы или отмена холинолитического корректора провоцируют развитие паркинсонизма. В клинической картине нейролептического паркинсонизма встречаются симптомы, не характерные для БП: окулогирные кризы, оральные гиперкинезы, тризм, тортиколис, кризы опистотонуса. Чаще симптоматика развивается с двух сторон. После отмены препарата происходит регресс симптоматики, обычно в течение нескольких недель. В некоторых случаях, особенно у пожилых пациентов, симптомы могут сохраняться годами.
Сосудистый паркинсонизм. Форма вторичного паркинсонизма, для которой характерна самая высокая частота гипердиагностики. По данным морфологических исследований, проводимых в специализированных центрах, частота сосудистого паркинсонизма не превышает 6–8 % всех случаев паркинсонизма.
Причины, вызывающие развитие сосудистого паркинсонизма, следующие:
1. Поражение мелких мозговых артерий:
— гипертоническая микроангиопатия (липогиалиноз);
— сенильная микроангиопатия (сенильный артериолосклероз, сенильная извитость артерий);
— васкулиты и васкулопатии (узелковый полиартериит, ангиит ЦНС, СКВ);
— наследственные артериопатии.
2. Поражение крупных мозговых артерий:
— атеросклероз крупных (экстра- и интракраниальных) артерий;
— менинговаскулярный сифилис.
3. Кардиогенные поражения головного мозга:
— кардиогенные эмболии;
— гипоксическая энцефалопатия (некроз базальных ганглиев).
4. Другие заболевания:
— артериовенозные мальформации;
— антифосфолипидный синдром;
— коагулопатии.
Морфологические изменения вещества головного мозга, обнаруживаемые при сосудистом паркинсонизме при нейровизуализации, следующие:
1) множественные лакунарные инфаркты в базальных ганглиях, стволе, глубинных отделах белого вещества;
2) диффузные поражения белого вещества:
— субкортикальный сливающийся или частично сливающийся лейкоареоз;
— распространенный перивентрикулярный лейкоареоз с неровными контурами, распространяющийся в субкортикальную область;
3) территориальные подкорковые инфаркты в базальных ганглиях и прилегающем белом веществе;
4) двусторонние (реже односторонние) территориальные инфаркты лобных долей;
5) одно-, двусторонние инфаркты в области таламуса;
6) геморрагические очаги в базальных ганглиях, среднем мозге, таламусе;
7) церебральная атрофия с расширением желудочковой системы и корковых борозд.
Для данной формы вторичного паркинсонизма характерно наличие пластической ригидности, которая сочетается с элементами пирамидной спастичности и преимущественным поражением нижних конечностей. Характерны симметричный акинетико-ригидный синдром, нарушения ходьбы по типу дисбазии, раннее появление постуральной неустойчивости. Тремор встречается крайне редко и не имеет типичного паркинсонического характера. Часто развивается псевдобульбарный паралич с явлениями дизартрии, дисфагии, насильственных эмоций. Характерны разница сухожильных рефлексов, наличие патологических рефлексов, мозжечковых нарушений. Достаточно быстро формируются когнитивные расстройства. Отмечается отсутствие эффекта от препаратов L-дофы. Однако следует отметить, что иногда небольшие дозы указанных препаратов приносят облегчение больным, что может быть связано с развивающейся с возрастом экстрапирамидной недостаточностью.
Все разнообразие клинических проявлений сосудистого паркинсонизма можно свести к трем наиболее характерным вариантам:
1. С симметричным началом в виде акинетико-ригидного синдрома, более выраженного в нижних конечностях, дебютирующего с нарушения ходьбы. Отсутствует тремор и эффект от препаратов L-дофы. Характерно постепенное развитие на фоне прогрессирующей сосудисто-мозговой недостаточности.
2. Характеризующийся сочетанием паркинсонизма с пирамидными, мозжечковыми, псевдобульбарными, когнитивными нарушениями, глазодвигательными расстройствами.
3. Сосудистый паркинсонизм с односторонним акинетико-ригидным синдромом, тремором покоя, иногда с позитивным ответом на препараты L-дофы. Вариант встречается при возникновении сосудистого поражения в среднем мозге, в области черной субстанции.
Для дифференциальной диагностики сосудистого паркинсонизма с БП необходимо использовать методы нейровизуализации (МРТ, ПЭТ).
Посттравматический паркинсонизм. Чаще всего развивается при тяжелой черепно-мозговой травме (ЧМТ) либо после повторных легких ЧМТ. После тяжелой ЧМТ паркинсонический синдром развивается в случае непосредственного травматического повреждения подкорковых ганглиев либо диффузного повреждения аксонов в глубинных отделах больших полушарий вследствие ротационного ускорения в момент травмы. При этом паркинсонизм развивается примерно в 1 % случаев и характеризуется гипокинезией, гипофонией, постуральной неустойчивостью и не носит прогрессирующего характера. При нейровизуализации часто обнаруживаются структурные изменения в подкорковых ганглиях.
Частые повторные легкие ЧМТ, особенно у людей, занимающихся контактными видами спорта (бокс, восточные единоборства), могут приводить к развитию энцефалопатии боксеров. Она включает в себя симптомы экстрапирамидных нарушений, обычно симметрично расположенных, а также пирамидные, мозжечковые, псевдобульбарные нарушения, развитие когнитивных расстройств. Считается, что причиной возникновения данной формы вторичного паркинсонизма являются мелкие повреждения в области ножек мозга, приводящие к аксональным нарушениям нигростриарного тракта.
Постинфекционный паркинсонизм. Впервые описан в 1918 году К. фон Экономо после перенесенного летаргического энцефалита. В качестве этиологического фактора рассматривали вирусную инфекцию (ортомиксовирус, вирус гриппа). Однако проведенные исследования не обнаружили в веществе головного мозга людей, умерших от летаргического энцефалита, РНК вируса гриппа. Это позволило предположить аутоиммунную природу заболевания, что было подтверждено в 2003 году исследователями из Великобритании, которые у 95 % пациентов со спорадическими случаями летаргического энцефалита в крови обнаружили аутоантитела к антигенам базальных ганглиев.
Клиническая картина заболевания развивается остро, начинается с повышения температуры тела, миалгий, общемозговой симптоматики. Кроме явлений паркинсонизма, наблюдаются специфические симптомы: выраженная гиперсомния, окулогирные кризы, глазодвигательные нарушения, приступы оцепенения или парадоксальной кинезии, дистонии, миоклонии, пирамидные знаки. Явления тремора преобладают над ригидностью. Характерны вегетативные нарушения: повышенная сальность кожи и волос, гиперсаливация, гипергидроз, различные вазомоторные нарушения. Течение заболевания может быть стационарным либо с медленным прогрессированием с нарастанием брадикинезии, ригидности, постуральных нарушений. Иногда наблюдается развитие деменции.
В настоящее время постэнцефалитический паркинсонизм наблюдается как следствие следующих инфекций:
— энтеровирусной (Коксаки типа В, поливирус);
— арбовирусной (клещевого и японского энцефалита);
— вирусов эпидемического паротита;
— вируса кори;
— вирусов гриппа типа А;
— вирусов простого герпеса;
— варицелла-зостерного вируса;
— ВИЧ;
— нейросифилиса;
— спирохетозной (Borrelia burgdorferi).
Токсический паркинсонизм. Может развиться вследствие воздействия различных экзогенных токсинов: марганца, оксида углерода, сероуглерода, таллия, цианидов, сероводорода, тетраэтилсвинца, ФОС, метанола. В последние десятилетия описаны случаи паркинсонизма у наркоманов при применении суррогатов героина, содержащего 1-метил-4-фенил-1,2,3,6-тетрагидроперидин (МФТП), который обладает высоким сродством к нейронам черной субстанции. Наиболее частая локализация поражения при токсическом паркинсонизме — бледный шар и черная субстанция.
Наиболее часто наблюдается паркинсонизм при интоксикации марганцем. Поступает марганец в организм при вдыхании пыли, содержащей его высокие концентрации, с пищей, парентерально (при использовании перманганата калия для приготовления наркотических препаратов). Заболевание развивается постепенно, в нем различают начальную и позднюю стадии. В ранней стадии на фоне неспецифической симптоматики (головная боль, астенизация, полиневропатии) обнаруживаются признаки акинетико-ригидного синдрома: повышение мышечного тонуса, мышечная дистония, тремор пальцев рук и век, гипомимия, гиперсомния, апатичность. Характерны вегетативные расстройства: тахикардия, артериальная гипотензия, гипергидроз, акроцианоз.
В поздней стадии заболевания прогрессируют акинетико-ригидные нарушения, которые сочетаются с пирамидными и церебеллярными расстройствами (гиперрефлексия, патологические рефлексы, интенционное дрожание, адаидохокинез и др.). Усиливаются полиневритические расстройства с нарастанием симптомов сенситивной атаксии, формируются астенодепрессивные реакции. Обратное развитие заболевания возможно только на ранней стадии.
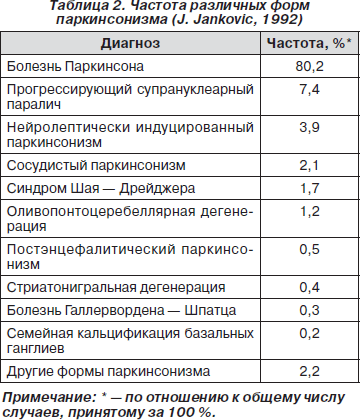
В табл. 2 представлена частота различных форм паркинсонизма. Следует обратить внимание на то, что первичный паркинсонизм встречается в подавляющем большинстве случаев и на его долю приходится около 80 % всех случаев паркинсонизма.
Основные принципы терапии БП
Как уже упоминалось выше, согласно концепции Braak и соавт., БП характеризуется восходящим типом нейродегенеративного процесса: от каудальных отделов ствола мозга к коре больших полушарий. При этом латентная и наиболее ранняя продромальная (премоторная) стадии болезни длятся около 5–8 лет и к моменту манифестации классических проявлений БП гибнет большая часть нейронов черной субстанции. Поэтому попытки нейропротекции на более поздней стадии не могут быть успешными, и задачей врача является вмешательство на максимально ранних стадиях патологического процесса.
Основными задачами лечения больных паркинсонизмом являются компенсация дофаминового дефицита и его последствий (коррекция нарушенных взаимоотношений нейромедиаторов), замедление прогрессирования заболевания (сохранение и защита дофаминовых нейронов), активация восстановительных процессов и стимуляция синтеза дофамина.
Для достижения поставленных задач необходимо придерживаться следующих принципов терапии БП:
— лечение назначается не позднее 6 месяцев от обращения пациента (срок точного установления правильного диагноза);
— при установлении диагноза БП больной должен непрерывно принимать противопаркинсонические средства (ППС) всю оставшуюся жизнь;
— лечение начинается в виде монотерапии одним из ППС;
— препаратом выбора может быть любое ППС (при отсутствии противопоказаний), в том числе холинолитик;
— монотерапия начинается с субпороговых доз (оптимальная доза подбирается постепенно, в течение не менее 3–4 недель) и осуществляется в пределах фармакотерапевтического окна, с установлением оптимальной однократной дозы и кратности приема;
— комбинированная терапия назначается при недостаточной эффективности монотерапии с добавлением второго ППС в субпороговой дозе с последующим постепенным увеличением дозы;
— появление побочных эффектов требует уменьшения дозы или отмены препарата, ответственного за побочный эффект, возможная утрата эффективности терапии компенсируется пересмотром доз и кратности приема других ППС;
— длительное лечение БП препаратами леводопы со временем приводит к возникновению клинического патоморфоза, требующего специальной коррекции.
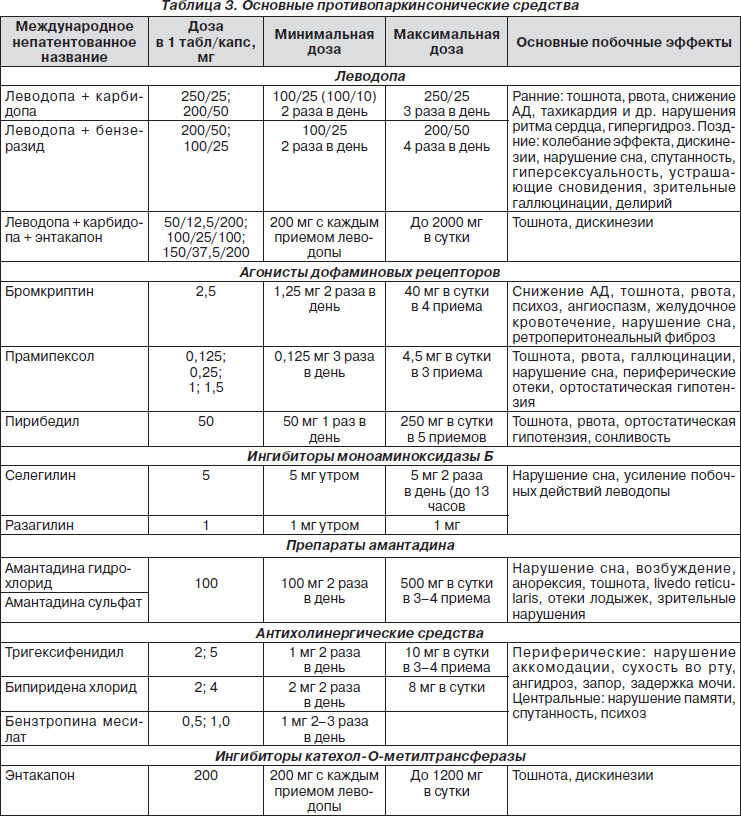
На практике сегодня применяются 6 основных групп ППС:
— препараты леводопы;
— агонисты дофаминовых рецепторов;
— ингибиторы ферментов метаболизма дофамина — КОМТ и МАО-Б;
— амантадины;
— центральные холинолитики.
Препараты леводопы (L-допа). Появились в клинической практике в 60–70-х годах XX столетия. Их использование позволило значительно отодвинуть сроки инвалидизации больных БП. L-допа является биологическим предшественником дофамина. Применение самого дофамина в качестве лечебного средства оказалось невозможным, так как он не проникает через гематоэнцефалический барьер. Однако существуют проблемы и с приемом L-допы, так как большая часть препарата разрушается в желудочно-кишечном тракте и крови под воздействием ДОФА-декарбоксилазы, что является причиной возникновения таких побочных эффектов, как тошнота, рвота, ортостатическая гипотензия. Лишь 1 % от принятой дозы леводопы достигает нигростриарных нейронов и принимает участие в синтезе дофамина. Для того чтобы нивелировать действие периферической ДОФА-декарбоксилазы и облегчить поступление леводопы в ткани мозга, уменьшить выраженность периферических побочных эффектов и снизить общую принимаемую дозу леводопы, ее обычно комбинируют с ингибитором периферической ДОФА-декарбоксилазы (карбидопа или бензеразид). В настоящее время препараты чистой леводопы практически не применяются в клинической практике. Использование препаратов L-допы считается золотым стандартом лечения БП.
Сроки начала терапии препаратами леводопы должны быть индивидуальными и зависят от темпа прогрессирования болезни, рода профессиональной деятельности, эффективности других ППС, семейно-бытового статуса пациента. Считается, что назначать препараты L-допы наиболее целесообразно тогда, когда паркинсонизм приводит к существенным двигательным нарушениям, которые не купируются другими ППС.
Выбор оптимальной дозы и кратности приема препарата зависит от индивидуальной чувствительности. Оптимальной индивидуальной дозой считается такая доза препарата, при которой наступает максимально возможная коррекция симптомов паркинсонизма, но не возникает побочных эффектов. Назначение дофасодержащих препаратов с ингибиторами ДОФА-декарбоксилазы начинается с субпороговых доз (табл. 3). В начале лечения назначают по 1/4 таблетки 3 раза в день, затем увеличивают дозу на 1/4 таблетки 1 раз в неделю. После каждого повышения дозы оценивают ее эффективность. Обычно при первом назначении леводопы достаточно принимать по 1/2 таблетки 3 раза в день. При необходимости дозу увеличивают до 1 таблетки 3 раза в день. Если дневная доза 4 таблетки не дает эффекта, дальнейшее увеличение дозы считается неэффективным.
Оценивать эффективность препарата можно с использованием простых тестов:
— теста пронации-супинации кисти;
— теста движения кисти между двумя точками, расположенными на расстоянии 30 см друг от друга;
— теста быстрых движений пальцев кисти (например, постукивания поочередно пальцами кисти по столу);
— теста ходьбы на расстояние 7 метров и обратно с поворотом туловища.
Следует отметить, что постепенное увеличение дозы препарата по мере нарастания степени тяжести заболевания оказывается неизбежным. Кроме того, с прогрессированием болезни уменьшается продолжительность действия разовой дозы, что вызывает необходимость увеличения кратности приема препарата. Однако с увеличением кратности приема уменьшается разовая доза препарата, что может привести к тому, что разовая доза может стать субпороговой и не оказывать клинического эффекта. В связи с этим кратность приема препарата не должна превышать 4–5 раз в сутки.
Использование ингибиторов ДОФА-декарбоксилазы позволило в большинстве случаев устранить периферические побочные действия леводопы. Однако у 60–80 % больных через 3–5 лет приема препаратов развиваются центральные побочные эффекты, такие как психотические эквиваленты, дискинезии, резкие флюктуации двигательной активности, на которых мы остановимся ниже.
Для достижения более стабильного уровня дофасодержащих препаратов в крови с целью устранения нефизиологичной, пульсирующей стимуляции дофаминовых рецепторов, уменьшения выраженности лекарственных дискинезий и времени их наступления используют препараты леводопы пролонгированного действия. Они обеспечивают замедленное высвобождение действующего вещества в ЖКТ, что способствует более длительному эффекту препарата (до 8 ч), однако относительно низкая биодоступность леводопы в составе данных лекарственных форм требует повышения общей дозировки действующего вещества в среднем на 30 %.
В начале 2000-х годов была предложена форма леводопы в сочетании с карбидопой для постоянного дуоденального введения после микрогастростомии. Благодаря переносной дозирующей помпе у больных, находящихся в тяжелой стадии болезни, обеспечивается постоянная концентрация леводопы в кишечнике и крови.
Из дофасодержащих ППС обращает на себя внимание зарегистрированный в Украине препарат Карбидопа и Леводопа-Тева (производитель — компания TEVA, Израиль). В его составе соотношение леводопы/карбидопы 10 : 1. Препарат представляет собой делимую таблетку, что позволяет оптимально дозировать прием препарата, начиная с 1/4 таблетки 3 раза в день, увеличивая дозу на 1/4 таблетки 1 раз в неделю. Препарат представляет собой идеальное сочетание цены и качества на фармацевтическом рынке.
Агонисты ДА-рецепторов. Первоначально агонисты ДА-рецепторов (АДАР) использовались для комбинированной терапии с препаратами леводопы, чтобы снизить их дозу. Впоследствии оказалось, что АДАР эффективны в начальных стадиях заболевания в качестве монотерапии. Особенно возрос интерес к препаратам данной группы после того, как в ряде исследований было показано нейропротективное действие АДАР, что замедляло темпы прогрессирования заболевания.
АДАР действуют непосредственно на ДА-рецепторы в подкорковых ганглиях, минуя пресинаптическую часть нигростриарных нейронов. Их действие направлено на D2-рецепторы, которые широко распространены в нигростриарных, мезолимбических и мезокортикальных путях. Именно со стимуляцией D2-рецепторов связан симптоматический эффект АДАР в отношении ригидности, гипокинезии и тремора. В настоящее время существует две группы АДАР: эрголиновые (бромкриптин) и неэрголиновые (пирибедил и прамипексол) (табл. 3).
К преимуществам АДАР относятся:
— эффективность в отношении тремора, плохо поддающегося традиционной терапии леводопой («антитреморный» эффект особенно убедительно показан для пирибедила и прамипексола);
— отсутствие конкуренции с пищевыми аминокислотами;
— отсутствие необходимости дальнейшего метаболизирования в ЦНС, в том числе с участием окислительных реакций;
— более длительный (по сравнению с леводопой) период полужизни и более длительная тоническая стимуляция постсинаптических рецепторов;
— меньший риск развития дискинезий;
— антидепрессивный эффект ряда препаратов (особенно у прамипексола);
— нейропротективное действие.
Среди побочных эффектов АДАР следует отметить:
— тошноту;
— рвоту;
— сердечные аритмии;
— фиброз клапанов сердца;
— постуральную гипотензию;
— галлюцинации;
— нарушение сна;
— периферические отеки;
— феномен Рейно и др.
В большей степени указанные побочные действия относятся к эрголиновым АДАР.
Помимо воздействия на D2-рецепторы, пирибедил усиливает центральную норадренергическую передачу за счет дополнительных a2-норадренергических свойств (блокада пресинаптических a2-адренорецепторов, реципрокное усиление высвобождения ацетилхолина в лобной коре и дорсальном гиппокампе). Благодаря этому эффекту пирибедил эффективен в лечении таких осложнений болезни Паркинсона, как когнитивные нарушения и постуральная неустойчивость.
При правильном индивидуальном подборе и постепенном повышении дозы АДАР показывают хорошую переносимость и достаточно высокую эффективность. На ранних стадиях БП, применяя в качестве монотерапии неэрголиновые АДАР, удается уменьшить степень выраженности основных симптомов заболевания (брадикинезия, тремор и мышечная ригидность) на 20–40 %, а у больных на развернутых стадиях заболевания добавление АДАР к леводопе способствует снижению тяжести симптоматики в среднем на 15–20 %. Около 60 % больных, получающих АДАР в качестве монотерапии, не нуждаются в назначении леводопы к концу третьего года лечения. Начальная монотерапия современными неэрголиновыми АДАР (с дальнейшим присоединением леводопы или без такового) сопровождается меньшей частотой возникновения дискинезии через 3–5 лет от начала лечения, причем качество жизни на фоне начальной монотерапии леводопой и АДАР практически одинаково. На поздних стадиях болезни при использовании АДАР в сочетании с леводопой отмечается сокращение до трети общей длительности периодов выключения, «сглаживаются» двигательные флюктуации, а также достигается возможность снижения общей суточной дозы леводопы на 25–30 %.
Ингибиторы моноаминоксидазы типа В (МАО-В). Фермент МАО типа В участвует в метаболизме церебральных моноаминов, в том числе дофамина, расщепляя его до конечного продукта — гомованилиновой кислоты. Кроме того, ингибиторы МАО-В являются антиоксидантами, защитное действие которых неоднократно показано на различных экспериментальных моделях паркинсонизма. Фармакотерапевтический эффект ингибиторов МАО-В связывают:
— с увеличением уровня дофамина в стриатуме;
— увеличением в стриатуме содержания фенилэтил-амина, который стимулирует высвобождение и тормозит обратный захват дофамина, а также его способностью стимулировать напрямую дофаминовые рецепторы;
— действием метаболитов селегилина — метамфетамина и амфетамина, которые усиливают высвобождение и тормозят обратный захват дофамина;
— способностью селегилина прямо или косвенно изменять активность дофаминовых нейронов, что приводит к повышенному высвобождению дофамина.
Суточная доза селегилина должна составлять 10–15 мг в сутки в 2 приема (табл. 3).
Относительно недавно стал применяться препарат нового поколения — необратимый ингибитор МАО-В разагилин. В проведенных исследованиях показана способность разагилина позитивно влиять на течение патологического процесса при БП, что позволяет отнести препарат к достаточно перспективным.
Ингибиторы катехол-О-метилтрансферазы (КОМТ). Фермент КОМТ осуществляет метилирование как предшественника дофамина L-допы, так и самого дофамина, которые перестают участвовать в осуществлении функций дофаминергических нейронов. Ингибиторы КОМТ способны повышать как уровень эндогенного дофамина, так и синтез дофамина из леводопы. Существуют ингибиторы КОМТ периферического действия, которые не проникают через гематоэнцефалический барьер (энтакапон), и проходящие через него (толкапон). Толкапон ввиду его высокой гепатотоксичности в качестве лечебного средства не применяется. Эффективная однократная доза энтакапона составляет 200 мг, среднесуточная доза — от 600 до 1200 мг. Препарат оказывает положительное влияние на моторные флюктуации, особенно при «изнашивании» конца дозы. Существует комбинированный препарат леводопа + карбидопа + энтакапон. Он позволяет облегчить борьбу с леводопа-индуцированными флюктуациями. Есть данные, что раннее назначение этой комбинации способно предотвращать либо отсрочивать наступление осложнений терапии леводопой (табл. 3).
Амантадины. Препараты из группы амантадинов достаточно давно используются для лечения БП. Известны две подгруппы амантадинов: амантадина гидрохлорид и амантадина сульфат. Эффективность в лечении БП связывают со следующими механизмами:
— увеличение синтеза дофамина в пресинаптических терминалях;
— увеличение выброса дофамина в синаптическую щель;
— торможение обратного захвата дофамина из синаптической щели;
— блокирование NMDA-рецепторов глутамата.
На ранних и среднетяжелых стадиях заболевания амантадины оказывают умеренный противопаркинсонический эффект, на развернутых стадиях они могут также уменьшать выраженность двигательных осложнений проводимой леводопа-терапии. Важным является свойство амантадинов подавлять выраженность индуцированных леводопой дискинезий. С учетом глутаматблокирующего эффекта амантадинов обсуждается целесообразность их назначения с целью коррекции имеющихся у больных когнитивных нарушений. Оптимальной дозой является 100–200 мг амантадинов в сутки в 3 приема (табл. 3).
Холинолитики. Препараты из группы холинолитиков применяются для лечения БП еще с середины XIX века. Механизм действия связан с восстановлением баланса между относительно преобладающей холинергической активностью и сниженной дофаминергической функцией в стриатуме. В настоящее время применение препаратов из группы холинолитиков снижается, что обусловлено достаточно большим количеством побочных эффектов. Из периферических побочных действий препарата следует отметить:
— нарушение аккомодации;
— мидриаз;
— сухость во рту;
— запоры;
— задержку мочеиспускания.
Из центральных побочных эффектов необходимо указать:
— галлюцинации;
— нарушение когнитивных функций.
Прямыми противопоказаниями для назначения холинолитиков являются аденома предстательной железы, глаукома, ряд форм сердечных аритмий, расстройства памяти и атрофические изменения головного мозга по данным нейровизуализации.
Назначение холинолитиков возможно в ранней стадии заболевания у относительно молодых пациентов (до 65 лет) в качестве монотерапии, главным образом при дрожательной форме БП. Комбинация с леводопасодержащими препаратами позволяет уменьшить выраженность моторных флюктуаций в течение дня, пролонгирует действие леводопы. Среднесуточная доза для большинства препаратов этой группы составляет 4– 8 мг в сутки (табл. 3).
Хирургическое лечение. На поздних стадиях заболевания, когда исчерпаны все возможности консервативной терапии и развиваются некупируемые двигательные нарушения, следует ставить вопрос о нейрохирургических методах лечения.
В настоящее время применяется два метода нейрохирургического воздействия на поздних стадиях БП:
— стереотаксическая деструкция определенных групп ядер таламуса, бледного шара и др.;
— хроническая высокочастотная электростимуляция глубоких структур мозга с использованием имплантированных электродов (функциональная нейрохирургия). Ее задача — прерывание патологически функционирующих паллидоталамокортикальных нейрональных «контуров».
Глубинная электростимуляция имеет преимущества перед деструктивными операциями: она может проводиться с двух сторон и характеризуется меньшим числом осложнений и более отчетливым эффектом в отношении всех основных клинических проявлений паркинсонизма.
Лекарственные дискинезии при БП
Как уже отмечалось, препараты леводопы являются золотым стандартом в терапии БП. Однако при длительной леводопатерапии наступает изменение типичной картины заболевания, ведущим проявлением которого являются лекарственные дискинезии, ухудшающие качество жизни пациентов.
Механизм развития дискинезий связан со следующими факторами:
— продолжающейся гибелью клеток черной субстанции;
— нефизиологичной, пульсирующей стимуляцией дофаминовых рецепторов;
— уменьшением активности медиального сегмента бледного шара и ретикулярной части черной субстанции.
Предложена следующая классификация дискинезий (J.A. Obeso, 1989):
1. Дискинезия (дистония) действия — проявляется в виде хореоформного гиперкинеза при определенном двигательном акте.
2. Дискинезия «включения» — возникает на фоне эффекта однократной дозы и подразделяется на дискинезию пика дозы, которая проявляется при максимальном эффекте препарата леводопы, а также дискинезию периода лечебного плато, которая прослеживается в течение всего периода «включения».
3. Двухфазная дискинезия появляется в начале периода «включения» и исчезает, с тем чтобы появиться в самом конце периода «включения». Указанная форма дискинезии чаще проявляется медленными дистоническими дискинезиями, обычно в мышцах ног. У части больных двухфазная дискинезия проявляется дистонией позы.
4. Дистония периода «выключения» появляется в период окончания действия однократной дозы леводопасодержащих препаратов. В основном проявляется в утреннее время в виде утренней дистонии после ночного перерыва в приеме препаратов. Обычно проявляется в виде дистонии позы, мышц лица и шеи, но может быть генерализованной.
5. Дискинезия-паркинсонизм проявляется возникновением гиперкинеза в одной половине тела с антипаркинсоническим эффектом, в другой половине тела — с симптомами паркинсонизма. В основе этого лежит различная чувствительность дофаминергических нейронов в соматотопически различных отделах стриатума.
6. Дискинезия без двигательного улучшения возникает после приема препаратов без какого-либо антипаркинсонического эффекта.
7. Пароксизмальная непредсказуемая дискинезия появляется в любое время независимо от времени приема препарата.
На ранних стадиях дискинезии можно скорректировать уменьшением дозы леводопы либо изменением кратности ее приема. При длительной терапии БП коррекция усложняется. При уменьшении дозировки препаратов либо кратности приема может ухудшаться двигательная активность пациентов: раннее появление симптомов «выключения» или большая выраженность этого периода.
Существует ряд групп препаратов, способных уменьшать выраженность и длительность дискинезии, при этом используются следующие подходы:
— оптимизация терапии агонистами дофаминовых рецепторов с одновременным уменьшением дозы леводопы;
— использование препаратов леводопы пролонгированного действия;
— использование в терапии ингибиторов КОМТ;
— увеличение частоты приема препаратов леводопы;
— использование в терапии больших доз амантадинов.
Существуют подходы к терапии в зависимости от вида дискинезий.
При наличии хореиформных дискинезий пика дозы можно использовать следующие методы:
— замена быстродействующих форм леводопы на пролонгированную форму;
— отмена ингибиторов МАО-В;
— уменьшение однократной дозы леводопы и/или увеличение дозы антагонистов дофаминовых рецепторов;
— назначение или увеличение дозы препаратов из группы амантадинов;
— нейрохирургическое лечение.
При наличии двухфазных дискинезий используют следующие методы коррекции:
— замена препарата леводопы пролонгированного действия на препарат быстрого действия;
— увеличение дозы препаратов дофаминергического действия;
— добавление ингибитора КОМТ;
— уменьшение утренней дозы леводопы и/или дневной дозы без изменения интервала между приемами;
— нейрохирургический метод.
При наличии дистонии раннего утра используется следующий подход к терапии:
— ночной прием препарата леводопы;
— ночной прием агонистов дофаминовых рецепторов;
— добавление ингибиторов КОМТ;
— утренний прием препаратов леводопы.
Перспективные методы лечения БП. В настоящее время большинство применяемых на сегодняшний день ППС относятся к группе симптоматических. Однако они не позволяют предотвратить или замедлить патологический процесс нейродегенерации либо вызвать его обратное развитие. В связи с этим в настоящее время разрабатывается и внедряется целый ряд новых противопаркинсонических средств, ориентированных не столько на симптоматический эффект, сколько на патогенетические основы болезни (рис. 2).
С учетом воздействия на патогенетические механизмы БП перспективным считается использование новых антиоксидантов (идебенон и др.), антагонистов глутаматных рецепторов, обладающих антиэксайтотоксическими свойствами (рилузол, ремасемид). Активно развивается направление, связанное с генной терапией БП, — стереотаксическое введение в область полосатого тела различных псевдовирусных наночастиц, несущих гены пептидных факторов роста, ферментов синтеза дофамина и т.д. Перспективы клеточной регенераторной терапии болезни Паркинсона зависят от того, насколько удачными будут попытки трансформации фенотипа применяемых клеток (стволовые мезенхимальные клетки костного мозга и жировой ткани, обкладочные клетки обонятельного эпителия и т.п.) по пути специфических ДА-продуцирующих нейронов.
Особые надежды возлагаются на разработку и клиническое внедрение новых молекулярных подходов к нейропротективной терапии нейродегенерации при БП:
— применение пептидных факторов роста — GDNF и др. путем интравентрикулярного введения;
— геннотерапевтические подходы к повышению экспрессии в мозге паркина, PINKI, DJ-1 и других белков;
— использование препаратов, повышающих экспрессию молекулярных шоперонов, активность протеасомного комплекса и интенсификацию процессов лизосомальной аутофагии;
— применение ингибиторов агрегации и процессинга a-синуклеина, ингибиторов киназной активности LRRK2;
— внедрение препаратов с цитокиновыми и противовоспалительными свойствами (эритропоэтин и др.), в том числе подавляющих реакции микроглиальной активации.
Однако для того чтобы новые методы терапии БП были эффективными, необходима их реализация на ранней или даже латентной стадии нейродегенеративного процесса, поскольку клинически манифестные формы БП связаны с гибелью 60–80 % дофаминергических нейронов. Это ставит задачу разработки адекватных биомаркеров БП — нейровизуализационных, нейрофизиологических, биохимических, молекулярно-генетических и др., которые были бы информативны в максимально ранние сроки болезни.


1. Болезнь Паркинсона и расстройства движений: Руководство для врачей / Под ред. С.Н. Иллариошкина, Н.Н. Яхно. — М., 2008. — 405 с.
2. Экстрапирамидные расстройства: Руководство по диагностике и лечению / Под ред. В.Н. Штока, И.А. Ивановой-Смоленской, О.С. Левина. — М.: МЕДпресс-информ, 2002. — 608 с.
3. Голубев В.Л. Лечение болезни Паркинсона: решенные и нерешенные вопросы // Избранные лекции по неврологии / Под ред. В.Л. Голубева. — М.: Эйдос Медиа, 2006. — С. 395–420.
4. Зальялова З.А., Яковлева Л.А., Богданов Э.И. Немоторные проявления болезни Паркинсона: Методическое пособие для постдипломного образования. — Казань, 2009. — 34 с.
5. Клиническая диагностика в неврологии: руководство для врачей / М.М. Одинак, Д.Е. Дыскин. — СПб.: СпецЛит, 2007. — 528 с.
6. Болезнь Паркинсона (этиология, патогенез, клиника, диагностика, лечение, профилактика) / Карабань И.Н., Карабань Н.В., Крыжановский Г.Н., Кучеряну В.Г., Магаева С.В. — М.: Медицина, 2002. — 336 с.