
Международный неврологический журнал 5 (43) 2011
Вернуться к номеру
Клинический случай нейрофиброматоза второго типа с множественными опухолями головного и спинного мозга
Авторы: Кушнир Г.М., д.м.н., профессор, заведующий кафедрой нервных болезней с курсом неврологии ФПО, Самохвалова В.В., к.м.н., ассистент кафедры нервных болезней с курсом неврологии ФПО, Крымский государственный медицинский университет им. С.И. Георгиевского, г. Симферополь
Рубрики: Неврология
Версия для печати
В статье представлены основные диагностические критерии нейрофиброматоза 1-го и 2-го типа. Особенностью описанного клинического случая пациента с нейрофиброматозом 2-го типа является наличие множественных опухолей головного и спинного мозга при практическом отсутствии кожных изменений и экстраневральной патологии.
Нейрофиброматоз, диагностические критерии, множественные опухоли центральной нервной системы.

 Нейрофиброматоз (НФ) — наследственное заболевание, предрасполагающее к развитию опухолей у человека [4, 5].
Нейрофиброматоз (НФ) — наследственное заболевание, предрасполагающее к развитию опухолей у человека [4, 5].
В литературе НФ 2-го типа (НФ 2) впервые описан в 1822 г. шотландским хирургом Wishart. НФ 1-го типа (НФ 1) был изучен в 1882 г. учеником Вирхова von Recklinghausen.
В 1916 г. Cushing в своей научной работе объединил эти заболевания под общим названием «болезнь Реклингхаузена». Однако после молекулярно-генетических исследований (результаты опубликованы в 1985 и 1987 гг.) были выявлены принципиальные отличия в патогенезе НФ 1 и НФ 2 и доказано, что это совершенно разные заболевания, требующие дифференцированного клинического подхода [3, 4, 6, 14, 15].
В литературе описано всего восемь типов нейрофиброматоза, однако в последнее время большинство из них (кроме НФ 2) считаются абортивными формами НФ 1 и в качестве самостоятельных нозологических форм не выделяются. Исключениями могут быть сегментарный нейрофиброматоз (НФ 5), когда типичные проявления НФ 1 локализуются в одном или нескольких соседних дерматомах (встречается крайне редко, обычно не наследуется), и не входящий в число восьми спинальный нейрофиброматоз, при котором симметрично поражаются все спинальные корешки (описано только несколько наблюдений) [6].
НФ 1 и НФ 2 являются аутосомно-доминантными генетическими заболеваниями без какого-либо расового или полового преобладания. Их локусы находятся соответственно на хромосомах 17q11.2 и 22q12.2 [3, 6, 14, 16]. Расположенные здесь гены кодируют синтез супрессоров опухолевого роста (белков нейрофибромина и мерлина), которые обеспечивают динамический контроль клеточного роста [13]. Наибольшее значение этот белок имеет в регулировании пролиферации клеток нейроэктодермального происхождения [14, 16].
При генетическом дефекте в соответствующих хромосомах динамическое равновесие регуляции роста смещается в сторону пролиферации и возникает доброкачественный опухолевый рост [13].
Для указанных заболеваний типична высокая частота спонтанных мутаций, в результате чего 50 % клинических случаев являются спорадическими. Оба заболевания характеризуются 100 % пенетрантностью и широкой фенотипической вариабельностью.
НФ 1 довольно распространен, его частота составляет примерно 1 : 3000. Частота НФ 2 равняется 1 : 40 000. Для обоих состояний характерна генетическая мозаичность [1, 4, 9].
Особый интерес для неврологов представляет нейрофиброматоз 2-го типа, который ранее называли центральным нейрофиброматозом и который предрасполагает к появлению доброкачественных новообразований в центральной нервной системе [12].
НФ 2, так же как и НФ 1, является аутосомно-доминантным заболеванием, однако встречается в популяции значительно реже [1].
Для НФ 2 характерны новообразования центральной и периферической нервной системы (чаще — шванномы) при минимальных кожных и экстраневральных симптомах. НФ 2 диагностируется у пациента при наличии какого-либо из нижеперечисленных симптомов:
1. Двусторонние новообразования 8-го черепного нерва, выявленные при помощи КТ или МРТ.
2. Наличие родственников 1-го порядка с НФ 2 и односторонним новообразованием 8-го нерва или 2 из нижеперечисленных заболеваний:
— глиома;
— менингиома;
— шваннома;
— нейрофиброма;
— ювенильное заднее подкапсулярное затемнение хрусталика.
НФ 1 характеризуется преимущественно кожными проявлениями (гиперпигментированными макулами цвета кофе с молоком, кожными и подкожными нейрофибромами), опухолями невральных оболочек (нейрофибромы), глиомами зрительного тракта и другими нейроонкологическими заболеваниями, целым рядом костных аномалий, когнитивным дефицитом и повышенным риском опухолевого роста за пределами нервной ткани [9, 10].
Средний возраст появления симптоматики при НФ 2 составляет 20 лет, средний возраст на момент постановки диагноза — примерно 28 лет. НФ 1, как правило, начинается в раннем детстве с кожных симптомов, тогда как НФ 2 — в молодом возрасте, чаще всего с развития глухоты в результате вестибулярных шванном (ВШ) или других признаков, вторичных относительно менингиом или спинальных шванном. Оба заболевания диагностируются на основании клинических признаков (табл. 1, 2).

Учитывая наличие множества неспецифических симптомов у больных, в 1988 году для диагностики НФ 2 Национальным институтом здоровья США разработаны абсолютные диагностические критерии (NIH criteria) [1, 5], а позже к ним добавлены вероятные критерии [11] (табл. 1).
3 % пациентов со шванномами и 1 % пациентов с менингиомами страдают НФ 2. 20 % пациентов с множественными менингиомами имеют НФ 2 [8, 11].
Наиболее характерное проявление НФ 2 — наличие двусторонних вестибулярных шванном [1, 4]. Вторые по частоте опухоли — это шванномы других черепных, спинальных и периферических нервов [2, 8]. Значительно реже (менее 10 %) встречаются менингиомы (интракраниальные, включая менингиомы зрительных нервов, и спинальные), эпендимомы и глиомы [2, 7].
В принципе, шванномы могут образовываться в любом месте в организме, где имеются нервы со шванновскими клетками. Излюбленная локализация опухолей на VIII нерве при НФ 2 до настоящего времени остается необъяснимой [4, 15, 16].
Чаще всего пациенты обращаются к врачу в связи со снижением слуха или с появленим шума в ушах, которые в начале заболевания носят односторонний характер. Данные жалобы могут сопровождаться головокружением и атаксией. В 20–30 % случаев у этих пациентов помимо вестибулярных шванном выявляют менингиомы, спинальные или периферические опухоли [2, 7].
Нередко заболевание манифестирует нейропатией лицевого нерва (3–5 %), которая не поддается лечению. У некоторых пациентов возникает полимиелитоподобный синдром (около 3 %) [15]. 60–80 % пациентов с НФ 2 имеют зрительные нарушения — катаракты, ретинобластомы, гемартромы, менингиомы зрительных нервов и другие [2, 7].
Приводим описание сложного клинического случая.
Пациент А., 26 лет, инвалид ІІ группы, обратился с жалобами на нарастающую слабость и нарушение чувствительности в ногах, больше правой; изменение походки, шаткость при ходьбе, усиливающуюся в темноте; императивные позывы на мочеиспускание; снижение слуха с обеих сторон; приступы выраженной головной боли, сопровождающиеся рвотой, чаще в утренние часы.
Из анамнеза известно, что заболевание началось около 6 лет назад, когда появились умеренные боли в поясничной области. 2 года назад присоединились вышеперечисленные жалобы. В последнее время беспокоят императивные позывы на мочеиспускание. Семейный анамнез не отягощен.
Обследовался в Киевском НИИ нейрохирургии, нейрохирургическом отделении КРУ «КБ им. Н.А. Семашко» г. Симферополя, в Турции. После проведения МРТ головного и спинного мозга был установлен диагноз: множественные объемные образования. Предлагалось оперативное лечение, от которого пациент отка- зался.
Объективно: общее состояние больного удовлетворительное. В соматическом статусе пациента патологии не выявлено.
Неврологический статус: сознание ясное, ориентирован, адекватен. Общемозговой и менингеальной симптоматики нет. Зрачки одинаковой величины, фотореакция и корнеальные рефлексы живые. Движения глазных яблок не ограничены. Речь и глотание не нарушены. Наблюдается снижение слуха с двух сторон, более выраженное слева. Лицо симметрично, язык по средней линии. Походка паретичная с элементом штампующей справа, выражена атаксия при ходьбе. Выявлен нижний спастический парапарез, более выраженный справа. Брюшные рефлексы отсутствуют. Выражена атаксия в позе Ромберга. Координаторные пробы выполняет с интенцией с двух сторон. Гипестезия по проводниковому типу с уровня D7 справа и с уровня L1 слева. Снижена глубокая чувствительность на нижних конечностях.
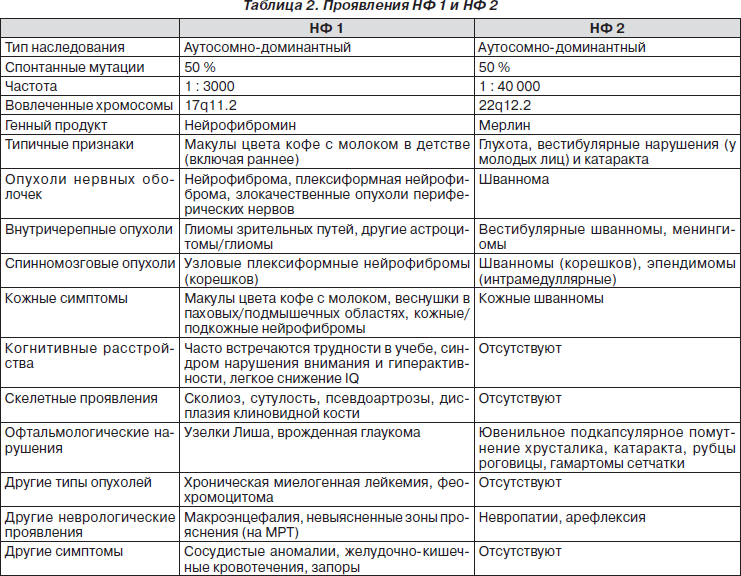
При осмотре больного обращает на себя внимание наличие множественных родинок на коже (рис. 1). Других кожных и подкожных образований не выявлено.
Дополнительные обследования: общие и биохимические анализы крови и мочи в пределах нормы.
Рентгенография органов грудной клетки и ЭКГ патологии не выявили.
На глазном дне: диски зрительных нервов бледно-розовые, границы стушеваны, с бледноватым оттенком, артерии сужены, извиты; вены темные, полнокровные, извиты. VOD = VOS = 1,0. Заключение: картина застойных дисков зрительных нервов обоих глаз.
Аудиометрия выявила поражение звуковоспринимающего аппарата с двух сторон. Заключение сурдолога: двусторонний базальный кохлеит.
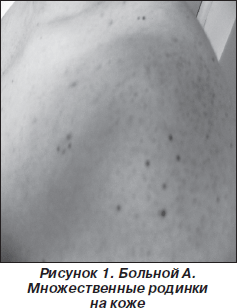
На МРТ головного мозга: множественные объемные образования гетерогенной структуры, содержащие кистозный компонент при контрастировании: в базальных отделах правой лобной доли размерами 21 х 25 х 19 мм, в области верхней лобной извилины размерами 47х 34 мм, в левой гемисфере мозжечка размерами 23 х 14 мм; билатерально от сильвиевой борозды подоболочечно выявлено образование, распространяющееся в глубь мозга, и в заднем роге правого бокового желудочка; эндо- и супраселлярное образование 35 х 15 мм, выполняющее турецкое седло и распространяющееся за его пределы, в мостомозжечковых углах и пирамидках на протяжении слуховых каналов двусторонние образования с деструкцией кости (рис. 2).
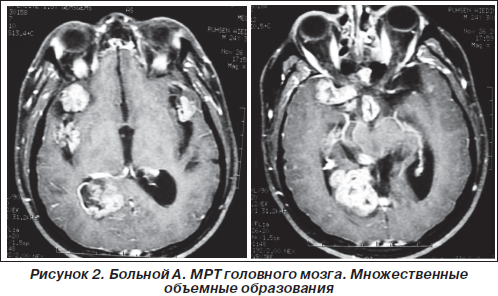
На МРТ шейного, грудного и пояснично-крестцового отделов позвоночника: на уровне С4 позвонка сирингомиелитическая полость размерами 18 х 13 х 27 мм; объемное образование в позвоночном канале на протяжении области исследования (С7-S5), накапливающее контраст. Спинной мозг прослеживается в виде отдельных фрагментов и оттеснен кпереди с уровня С7 до L1 (рис. 3).
Учитывая молодой возраст пациента, клиническую манифестацию заболевания в 20-летнем возрасте, двустороннее снижение слуха, наличие множественных опухолей в головном (в том числе и слуховых нервов) и спинном мозге на МРТ, минимальные кожные изменения и отсутствие экстраневральной патологии, был установлен диагноз: нейрофиброматоз 2-го типа с множественными опухолями головного и спинного мозга. Нижний спастический парапарез, смешанная атаксия. Нарушение функции тазовых органов по центральному типу. Ликворно-гипертензионные кризы.
Обсуждение
Следует отметить, что НФ 2 встречается значительно реже, чем НФ 1. Подобный клинический случай в неврологической практике можно встретить нечасто.
Диагностика НФ 2 весьма затруднительна из-за практического отсутствия внешних изменений (пятна на коже, опухоли) и неспецифичности неврологической картины.
У нашего пациента имелись множественные новообразования центральной нервной системы. В связи с тем, что оперативное лечение и биопсия опухолей не проводились, можно лишь предполагать наличие двусторонних шванном слуховых нервов. Описанное на МРТ объемное образование в позвоночном канале, оттесняющее спинной мозг, можно трактовать как множественные шванномы спинномозговых корешков. Также у больного не было кожных изменений, что характерно для НФ 2, за исключением многочисленных мелких родинок.
Следует отметить отсутствие когнитивных нарушений, патологии скелета, поражения внутренних органов, что типично для НФ 2. В то же время отсутствовали офтальмологические расстройства (ювенильное подкапсулярное помутнение хрусталика, катаракта, рубцы роговицы, гамартомы сетчатки), характерные для НФ 2.
В заключение следует отметить, что НФ является комплексной генетической патологией с множественными признаками и значительной фенотипической вариабельностью. НФ 2 ограничен, как правило, нервной системой, тогда как НФ 1 является системным расстройством. Сложность диагностики и лечения данных заболеваний требует координированного междисциплинарного подхода.
1. Asthagiri A.R., Parry D.M., Butman J.A. et al. Neurofibromatosis type 2 // Lancet. — 2009. — Vol. 6. — P. 1974-86.
2. Evans G.R., Watson C., King A. et al. Multiple meningiomas: differential involvement of the NF2 gene in children and adults // J. Med. Genet. — 2005. — Vol. 42. — P. 45-48.
3. Farrell C.J., Plotkin S.R. Genetic causes of brain tumors: neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes // Neurol. Clin. — 2007. — Vol. 25. — P. 925-46.
4. Ferner R.E. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective // Lancet Neurol. — 2007. — Vol. 6. — P. 340-51.
5. Gerber P.A., Antal A.S., Neumann N.J. et al. Neurofibromatosis // Eur. J. Med. Res. — 2009. — Vol. 14. — P. 102-5.
6. Gottfried Oren N., Viskochil David H., Fults Daniel W. et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications // Neurosurgery. — 2006. — Vol. 58. — P. 1-16.
7. Hartmann C., Sieberns J., Gehlhaar C. et al. NF2 mutations in secretory and other rare variants of meningiomas // Brain Pathol. — 2006. — Vol. 16. — P. 15-9.
8. Holland K., Kaye A.H. Spinal tumors in neurofibromatosis-2: management considerations — a review // J. Clin. Neurosci. — 2009. — Vol. 16. — P. 169-77.
9. Hottinger A.F., Khakoo Y. Neuro-oncology of Neurofibromatosis Type 1 // Curr. Treat. Options Neurol. — 2009. — Vol. 11. — P. 306-14.
10. Lee M.J., Stephenson D.A. Recent developments in neurofibromatosis type 1 // Curr. Opin. Neurol. — 2007. — Vol. 20. — P. 135-41.
11. McClatchey A.I. Neurofibromatosis // Annu. Rev. Pathol. — 2007. — Vol. 2. — P. 191-216.
12. Nowak C.B. The phakomatoses: dermatologic clues to neurologic anomalies // Semin. Pediatr. Neurol. — 2007. — Vol. 14. — P. 140-9.
13. Otibi M., Rutka J.T. Neurosurgical implications of neurofibromatosis Type I in children // Neurosurg Focus. — 2006. — Vol. 20. — P. 130-9.
14. Savar A., Cestari D.M. Neurofibromatosis type I: genetics and clinical manifestations // Semin. Ophthalmol. — 2008. — Vol. 23. — P. 45-51.
15. Williams V.C., Lucas J., Babcock M.A. et al. Neurofibromatosis type 1 revisited // Pediatrics. — 2009. — Vol. 123. — P. 124-33.
16. Yohay K. Neurofibromatosis type 1 and associated malignancies // Curr. Neurol. Neurosci. Rep. — 2009. — Vol. 9. — P. 247-53.