
Международный неврологический журнал 7 (45) 2011
Вернуться к номеру
Клинико-томографические соотношения при синдроме Арнольда — Киари
Авторы: Латышева В.Я., Олизарович М.В., Филюстин А.Е., Гурко Н.А., Учреждение образования «Гомельский государственный медицинский университет», Республиканский научно-практический центр радиационной медицины и экологии человека, г. Гомель, Республика Беларусь
Рубрики: Неврология
Версия для печати
Изучен 91 случай опущения миндалин мозжечка ниже края большого затылочного отверстия. Установлены характерные жалобы пациентов и неврологические расстройства в группах с изолированным и сочетанным синдромом Киари. Выявлены и подтверждены на МРТ нозологические формы, наиболее часто сочетающиеся с этой врожденной мальформацией.
Аномалия Арнольда — Киари, клиника, МРТ.

Введение
В последние десятилетия отмечается повышенный интерес к краниовертебральной патологии, при которой наблюдаются очаговые неврологические органические знаки, нарушение ликвородинамики, сосудистые нарушения головного и спинного мозга.
Краниовертебральные аномалии — это врожденные или приобретенные дефекты развития краниовертебрального перехода структур головного и спинного мозга или костных структур основания черепа и двух верхних шейных позвонков (платибазия, базилярная импрессия, атлантоаксиальный подвывих, ассимиляция атланта), которые служат основанием диагностики первичного или вторичного генеза клинических проявлений.
Опущение миндалин мозжечка может быть врожденным (первичным) или приобретенным (вторичным) в результате частых люмбальных пункций или после люмбоперитонеального шунтирования.
Аномалия (порок, мальформация) Киари — врожденное нарушение строения мозга, которое характеризуется низким расположением миндалин мозжечка. Заболевание названо в честь австрийского патолого-анатома Ханса Киари (Hans Chiari), который в 1891 году описал несколько типов аномалий развития ствола мозга и мозжечка.
Синдром Арнольда — Киари представляет церебелло-медуллярное уродство, которое характеризуется врожденным опущением продолговатого мозга и миндалин мозжечка в суженное большое затылочное отверстие. Эта мальформация характеризуется внутричерепной гипертензией, стволовыми нарушениями, каудальной дислокацией миндалин мозжечка и платибазией, при которой происходит вдавление основания затылочной кости и ската в заднюю черепную ямку [1]. Платибазия — это уплощение основания черепа, в результате чего скат расположен более горизонтально по отношению к плоскости передней черепной ямки. Платибазия клинически не проявляется, но в сочетании с другой костной аномалией черепа может вызывать различные симптомы. Сдавление спинного мозга приводит к двигательным нарушениям (спастическим парапарезам), а ствола мозга — к вовлечению в патологический процесс V–XII пар черепных нервов и нередко сопровождается зрительными нарушениями.
Современная патоморфология выделяет три основных типа этой аномалии: I — опущение миндалин мозжечка через большое затылочное отверстие в шейный отдел позвоночного канала. Этот тип мальформации может сопровождаться гидромиелией (расширением центрального канала спинного мозга) и костными краниовертебральными аномалиями. Обычно проявляется в подростковом или взрослом возрасте на третьем-четвертом десятилетии жизни.
Жалобы пациентов при I стадии включают боль в шейно-затылочной области при кашле, натуживании, нередки падения вследствие кратковременной ишемии каудального отдела спинного мозга и внезапного снижения мышечного тонуса по типу дроп-атак, обмороков, апноэ во сне. Нередко развиваются ликвородинамические нарушения и внутричерепная гидроцефалия, определяется бьющий вниз нистагм.
Клинические проявления аномалии Киари I складываются из различных сочетаний симптомов сдавления продолговатого мозга, сирингомиелии и гидроцефалии и обусловлены компрессией нижних отделов ствола, верхнешейных спинномозговых корешков этого отдела спинного мозга, мозжечка, черепных нервов каудальной группы, позвоночных артерий [5].
В. Williams объясняет начало клинических проявлений заболевания затруднением оттока ликвора из полости черепа в спинальное субарахноидальное пространство вследствие сужения большой затылочной цистерны при опускании миндалин мозжечка. Это приводит к повышению интракраниального и снижению интраспинального ликворного давления [3].
Самым частым симптомом является головная боль. Особенно характерна боль в затылочной области, усиливающаяся при покашливании и натуживании, боли в шее, слабость и нарушение чувствительности рук, неустойчивая походка, диплопия, смазанная речь, затрудненное глотание, рвота и шум в ушах.
При аномалии Киари низко расположенные миндалины мозжечка затрудняют свободную циркуляцию спинномозговой жидкости между головным и спинным мозгом. Миндалины блокируют большое затылочное отверстие, в результате чего нарушается отток ликвора и развивается гидроцефалия.
Мальформация Киари II называется также мальформацией Арнольда — Киари. Данное заболевание было названо в честь немецкого патологоанатома Юлиуса Арнольда, который более подробно описал данное заболевание в 1894 г. Частота его составляет от 3,3 до 8,2 наблюдений на 100 тыс. населения.
При II типе мальформации происходит вклинение дисплазированного мозжечка (червя и миндалин) вниз в большое затылочное отверстие в сочетании с удлинением ствола головного мозга и четвертого желудочка. При этой патологии часто определяется сочетание сирингомиелии, незаращения дужек позвонков (spina bifida) в пояснично-крестцовом отделе, спинномозговой грыжи, стеноза сильвиева водопровода с развитием гидроцефалии. На первом году жизни заболевание проявляется приступами удушья, остановки дыхания, развитием аспирационной пневмонии, что нередко приводит к летальному исходу.
Тип Арнольда — Киари III заключается в смещении мозжечка и части ствола мозга в менингоцеле, расположенное в шейно-затылочной области. Эти изменения сопровождаются субокципитальным или высоким шейным энцефаломенингоцеле и обычно несовместимы с жизнью.
При аномалии Арнольда — Киари III наблюдается врожденное увеличение диаметра большого затылочного отверстия. В сочетании возможны также гидроцефалия, сердечно-сосудистые аномалии, атрезия заднего прохода и другие нарушения пищеварительного тракта, нарушение развития мочеполовой системы. Жалобы при этом типе мальформации включают головокружение или шаткость при ходьбе (может усиливаться при повороте головы); шум (звон, гул, свист, шипение) в одном или обоих ушах; головную боль, связанную с повышением внутричерепного давления (более выраженную утром) или с повышением тонуса мышц шеи.
Мальформация Киари IV типа сопровождается недоразвитием мозжечка, заключается во врожденной гипоплазии мозжечка и в классификацию не включена. Эта патология приводит к летальному исходу младенца.
Патология краниоспинального перехода чаще выявляется при компьютерной или магнитно-резонансной томографии (КТ, МРТ). Наиболее распространены аномалии Арнольда — Киари I и II типа.
Патогномоничным признаком аномалии является опускание миндалин мозжечка более чем на 3–5 мм ниже плоскости большого затылочного отверстия.
Наряду с общепринятыми методиками выявления данной патологии при лучевой диагностике и МРТ-исследовании у взрослых возможна ранняя ультразвуковая диагностика мальформации у плода в сроках гестации от 22 до 28 недель. Используется горизонтальная и фронтальная плоскости сканирования, при этом выявляемость признаков мальформации Киари составляет от 24 до 45 % [3].
В связи с вышеизложенным представляет интерес проведение ранней диагностики у пациентов с этой патологией и определение тактики лечения.
Цель исследования — сравнительная оценка изолированных (первичных) и сочетанных (вторичных) форм мальформации Арнольда — Киари в Гомельской области за последние 10 лет и раннее выявление манифестации клинических симптомов заболевания.
Материал и методы
Проведен анализ 91 случая обращения жителей г. Гомеля и Гомельской области в Республиканский научно-практический центр радиационной медицины и экологии человека, у которых при МРТ-исследовании головного мозга выявлено пролабирование миндалин и/или полушарий мозжечка в большое затылочное отверстие. Пациенты разделены на две группы: первая (основная) группа включала пациентов с врожденной мальформацией Киари (84 чел.), вторую (группу сравнения) мы отнесли к синдромальной — с сочетанием пролабирования миндалин мозжечка вследствие опухоли и водянки головного мозга (7 чел.). Проанализированы следующие показатели: половозрастная структура, характер мальформации головного мозга в сочетании с сопутствующей мозговой патологией и степень влияния ее на клиническую картину.
Методом исследования было сопоставление и анализ этиологических и клинических данных, заключений МРТ в двух группах пациентов.
Результаты и обсуждение
Полученные данные по возрастному составу представлены в табл. 1.
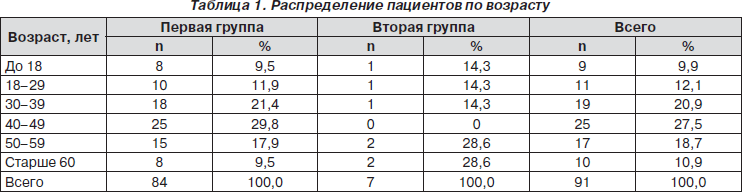
Как следует из табл. 1, группы характеризовались превалированием числа пациентов среднего и старшего возраста (старше 40 лет). В первой группе было 84 (57,2 %) чел., во второй — 7 (57,2 %) пациентов.
Гендерные особенности групп представлены в табл. 2.
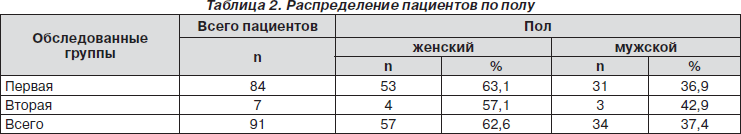
Данные табл. 2 свидетельствуют о значительном превалировании женщин в первой группе (53 чел. — 63,1 %), при несущественной разнице по половому признаку – во второй.
Характеристика неврологических нарушений при поступлении в стационар
При обращении пациентов в клинику изучали жалобы, анамнез заболевания, проводили неврологическое обследование по общепринятой методике и выполняли МРТ головного мозга.
При оценке неврологического статуса основное внимание уделяли клиническим проявлениям поражения мозжечка и ствола головного мозга. Диагноз устанавливали с учетом данных томографии по ранее изложенным критериям [5]. Среди пациентов первой группы (84 чел.) тип Киари I определялся у 78 (92,9 %) чел., тип Киари II — у 6 (7,1 %).
Наиболее часто встречающиеся жалобы у пациентов обеих групп по частоте выявления приведены в табл. 3.
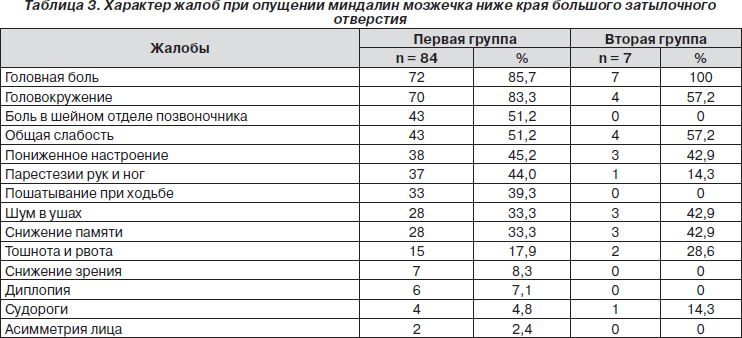
Результаты исследования, представленные в табл. 3, свидетельствуют о том, что наиболее частыми жалобами у пациентов с врожденной мальформацией Киари были головная боль и головокружение, часто в сочетании (соответственно 72 чел. — 85,7 % и 70 чел. — 83,3 %). Во второй группе у всех обследованных отмечались головная боль, головокружение и общая слабость. Несколько реже пациенты жаловались на боль в шейном отделе позвоночника, снижение настроения, парестезии в руках и ногах, пошатывание при ходьбе.
Данные, полученные при неврологическом обследовании пациентов, в ряде случаев позволили установить топический диагноз до выполнения томографического исследования. Результаты исследования неврологического статуса представлены в табл. 4.
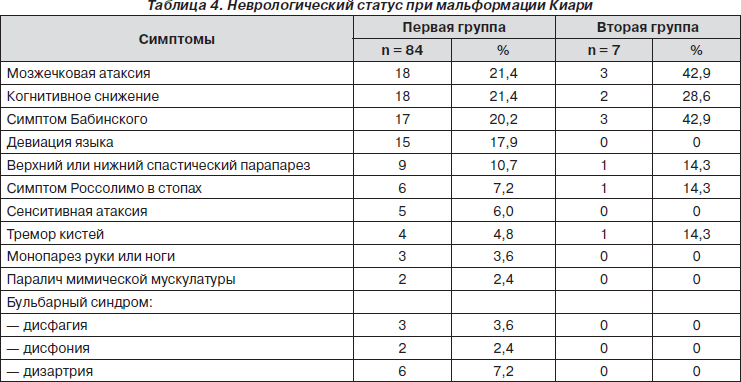
Как следует из табл. 4, наиболее частыми неврологическими симптомами при синдроме Киари были мозжечковая атаксия и когнитивное снижение (по 18 чел. — 21,4 %), во второй группе — мозжечковая атаксия и патологический симптом Бабинского с двух сторон (по 3 чел. — 42,9 %).
Основным методом достоверности диагноза была МРТ. Результаты МРТ-исследования свидетельствуют о взаимоотношении клиники и результатов томографии.
Приводим выписку заключения МРТ-исследования головного мозга по протоколам сканирования Sag T1, Ax Flair, Cjr T2, DW-EPI пациента А., 1954 г.р. (от 11.04.2011): «Объемных образований в веществе головного мозга не определяется. Определяется ретроцеребеллярная киста р. около 6 5 см, четвертый желудочек компремирован. Желудочковая система расширена: боковые — 25 мм, третий – 10 мм. Дислокации срединных структур нет. Вокруг боковых желудочков – повышенный в Flair сигнал. Структуры селлярной области без особенностей. Миндалины мозжечка пролабируют в затылочное отверстие до 5 мм. Околоносовые пазухи пневматизированы. Заключение: синдром Киари. Окклюзионная внутренняя гидроцефалия. Ретроцеребеллярная киста».
Однако следует отметить, что не всегда выраженная компрессия ствола головного мозга соответствовала неврологическому дефициту. Можно полагать, что компрессия продолговатого мозга зависела от величины большого затылочного отверстия. В некоторых случаях при пролабировании миндалин мозжечка отмечалась только головная боль без неврологического дефицита (рис. 1).
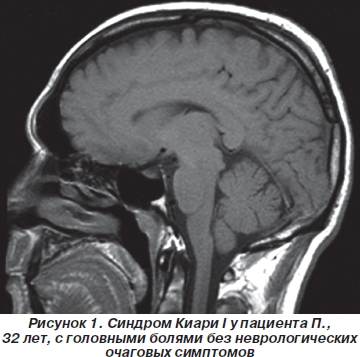
МРТ пациента, у которого опущение миндалин мозжечка в большое затылочное отверстие сопровождалось легким пошатыванием при ходьбе и тремором правой руки, представлена на рис. 2.
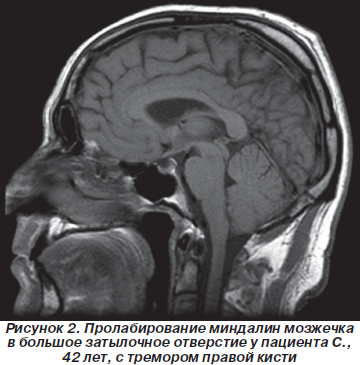
У лиц молодого возраста пошатывание при ходьбе, изменение походки вызывали затруднения при проведении дифференциальной диагностики. Так, через 3 года после начала заболевания обратился пациент 18 лет, который был направлен в клинику с диагнозом «демиелинизирующее заболевание центральной нервной системы. Подозрение на рассеянный склероз». При МРТ-обследовании у пациента, который жаловался на общую слабость, пошатывание при ходьбе, при обследовании определялся неврологический дефицит только в виде мозжечковой атаксии (рис. 3).
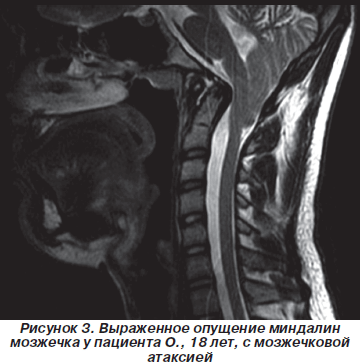
Полученные результаты МРТ-исследования позволили исключить демиелинизирующее заболевание центральной нервной системы. При анализе томограмм головного мозга была установлена следующая патология, с которой чаще всего сочеталась аномалия Арнольда — Киари. Результаты исследования представлены в табл. 5.
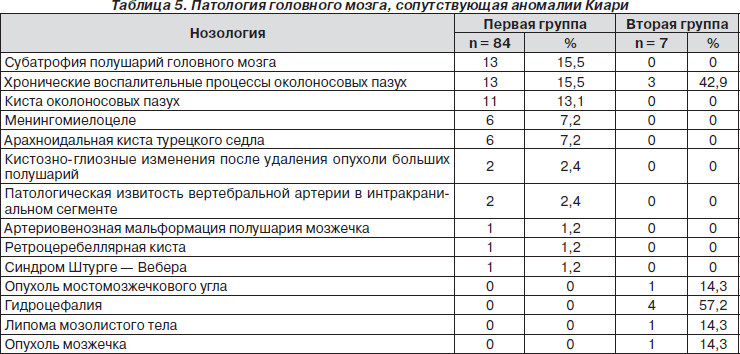
Представленный в табл. 5 анализ результатов исследования свидетельствует о том, что в первой группе как сопутствующие патологии головного мозга наиболее часто встречались субатрофия больших полушарий головного мозга и хронический воспалительный процесс околоносовых пазух (по 13 случаев — 15,5 %), менингомиелоцеле и синдром пустого турецкого седла (по 6 случаев — 7,2 %). Обращает на себя внимание значительное число пациентов с поражением околоносовых пазух, включая хроническое воспаление и кисты.
Во второй группе превалировали 4 случая гидроцефалии (57,2 %) и 3 – хронического гайморита (42,9 %). У некоторых пациентов определялись сочетания выявленной патологии.
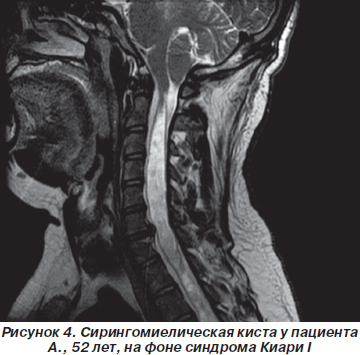
Результаты МРТ-исследования пациента А., у которого на фоне синдрома Киари I визуализировалась сирингомиелическая киста шейного и грудного отдела спинного мозга, представлены на рис. 4.
Объемных образований в веществе головного мозга не определяется. Определяется ретроцеребеллярная киста размером около 6 5 см, четвертый желудочек компремирован. Желудочковая система расширена: боковые — 25 мм, третий — 10 мм. Дислокации срединных структур нет. Вокруг боковых желудочков — повышенный в Flair сигнал. Структуры селлярной области без особенностей. Миндалины мозжечка пролабируют в затылочное отверстие до 5 мм. Околоносовые пазухи пневматизированы. Заключение: синдром Киари. Окклюзионная внутренняя гидроцефалия. Ретроцеребеллярная киста.
Дифференциальная диагностика синдрома Киари нами проводилась с большой группой заболеваний (16 чел.), для которых были характерны головные боли, атаксия, головокружение, тетрапарез, бульбарные нарушения. Чаще всего это были: опухоли задней черепной ямки или краниоспинального перехода; рассеянный склероз; рефлекторные, корешковые синдромы, синдром заднего шейного симпатического узла при шейном остеохондрозе; сосудистые нарушения ствола мозга при вертебробазилярной недостаточности; патологические изменения основания черепа (платибазия).
Представленный перечень свидетельствует о клинической значимости врожденной аномалии, которая может манифестировать различными заболеваниями и затруднять выбор патогенетической терапии.
Выводы
1. Врожденная аномалия Арнольда — Киари наиболее часто диагностировалась в среднем и старшем возрасте (старше 40 лет — 57,2 %), преимущественно у лиц женского пола (63,1 %).
2. Наиболее частыми жалобами у пациентов с врожденной мальформацией Арнольда — Киари были головная боль (85,7 %) и головокружение (83,3 %), основным неврологическим проявлением — мозжечковая атаксия и когнитивное снижение (по 21,4 %).
3. Характерная сопутствующая патология при синдроме Киари представлена субатрофией больших полушарий головного мозга (15,5 %), менингомиелоцеле и синдромом пустого турецкого седла (по 7,2 % случаев).
1. Скоромец А.А., Скоромец А.П., Скоромец Т.А. Нервные болезни: Учеб. пособие. — М.: МЕДпресс-информ, 2008. — С. 231-258.
2. Gardner W.J. Hydrodynamic mechanism of syringomielia: Its relationship to mielocele // J. Neurol. Neurosurg. Psychiat. — 1965. — Vol. 28. — P. 247-259.
3. Williams B. Simultaneous cerebral and spinal fluid pressure recordings. Cerebrospinal dissociation with lesions at the foramen magnum // Acta Neurochir. (Wien). — 1981. — Bd. 59. — S. 123-142.
4. Itoh Y., Kuwahara N., Sasajima Т., Mizoi K., Hatasawa J. Spinal cord edema preceding syringomyelia associated with Chiari I malformation case report // Neurologia medico-chirurgica (Tokyo) Neurol Med Chir (Tokyo). — 2002, Sep. — 42(9). — PP. 4103.
5. Крупина Н.Е. Сведения о семейных случаях сирингомиелии, базилярной импрессии и мальформации Киари // Неврологический вестник. — 2001. — № 1–2. — С. 70-75.
6. Менделевич Е.Г., Михайлов М.К., Богданов Э.И. Сирингомиелия и мальформация Арнольда — Киари. — Казань: Медицина, 2002. — 236 с.