
Журнал «Вестник Ассоциации психиатров Украины» (01) 2012
Вернуться к номеру
Синдром Ретта у мальчика. Презентация случая
Авторы: В.И. Харитонов, заведующий детским психоневрологическим отделением № 11 Киевской городской клинической психоневрологической больницы № 1; А.И. Кашин, генетик клиники «Евролаб»
Рубрики: Психиатрия
Версия для печати
Синдром Ретта — заболевание, относящееся к группе нарушений общего развития. Заболевание поражает практически только девочек (описано 60 случаев болезни у мальчиков), характеризуется нормальным ранним развитием и ростом, за которым следует замедление развития, утрата произвольного движения рук, появление стереотипий рук, замедление темпов роста головного мозга и головы, развитие проблем походки, появление судорог, а также интеллектуального дефицита. Синдром был впервые описан Андреасом Реттом в 1966 году [1], однако до 1983 года это заболевание не было признанным, и только с публикации Бенгта Хагберга в 1983 году началось активное изучение заболевания по всему миру. Причина состояния — мутация гена MECP2, локализованного на Х-хромосоме. В 10 % случаев заболевание является следствием мутации гена CDKL5 или FOXG1. В ряде случаев мутацию так и не удается определить, поэтому заболевание диагностируется в первую очередь клинически. В 95 % случаев синдром Ретта возникает вследствие мутации, возникшей de novo, т.е. родители при этом генотипически нормальные, без мутации. В спорадических случаях мутация обычно передается от мужской копии Х-хромосомы [2]. Пока не понятно, что заставляет мутировать сперму.
Ген МЕСР2 кодирует протеин МеСР2, который регулирует экспрессию других генов, подавляя их функции. Недостаточное функционирование протеина позволяет другим генам проявлять свои качества без какой-либо организации, что приводит к нарушению созревания головного мозга — снижению роста объема полушарий головного мозга и ствола, а также к нарушению их функционирования [3].
Критерии диагностики [4]:
— нормальный пре- и перинатальный анамнез;
— психомоторное развитие нормальное до 6 месяцев либо может быть задержано с рождения;
— нормальная окружность головы с постнатальной задержкой темпов развития (у большинства);
— потеря навыков движения руками;
— стереотипии рук: в виде мытья рук, потирания, хлопанья, засовывания рук в рот;
— нарушенные локомоторные функции (диспраксия).
Дополнительные критерии:
— расстройства дыхания во время бодрствования (гипервентиляция, задержки дыхания, глотание воздуха);
— бруксизм;
— нарушение паттерна сна (с раннего детства);
— нарушение мышечного тонуса;
— периферические вазомоторные расстройства;
— сколиоз/кифоз;
— задержка соматического роста;
— маленькие холодные кисти и стопы;
— периоды вскрикивания/смеха, не соответствующие ситуации.
Данный синдром также имеет определенную стадийность [5]:
— стадия 1 — пререгрессивный период (с 6–18 мес.): симптомы заболевания мало заметны — уменьшение контакта глазами, снижение интереса к игрушкам;
— стадия 2 — регрессивный период (1–4 года): потеря навыков, аутистическое поведение;
— стадия 3 — псевдостабилизационный период (4–10 лет): регресс не наблюдается, появляются атаксия, спастика и эпилепсия;
— стадия 4 — неамбулантная (15–25 лет): ухудшение моторных функций, паркинсонизм, улучшение коммуникаций, возможно улучшение функции рук и ходьбы.
В 11-е отделение Киевской городской клинической психоневрологической больницы № 1 поступил мальчик в возрасте 3 года 2 месяца. Родители обратились с жалобами на отсутствие речи, снижение интеллекта, частые автоматизмы (облизывает руки), бруксизм, шаткость во время ходьбы, приступы учащенного дыхания, эпилептические припадки. Ребенок от второй беременности, вторые роды (первая беременность закончилась рождением мальчика, которому сейчас 6 лет, и он является здоровым). Данная беременность протекала нормально. Роды в 38 недель. Вес при рождении 3000 г, рост 49 см, окружность головы не была указана в выписке. Оценка по шкале Апгар была 8/8 баллов. Домой мальчик был выписан на третьи сутки с диагнозом «здоров». До восьми месяцев ребенок развивался нормально. В восемь месяцев после перенесенного двустороннего перфоративного отита начал терять приобретенные навыки (перестал играть в ладушки, махать рукой на прощание). С восьми месяцев до одного года трех месяцев постепенно терял речь (в восемь месяцев говорил «дай», «ма», «па»), исчезли целенаправленные, произвольные движения руками. В возрасте два года и два месяца после прохождения курса цереброкурина у мальчика появились эпилептические припадки по типу простых и сложных парциальных миоклоний. Также в два года и два месяца у ребенка появились стереотипии в руках по типу облизывания рук.
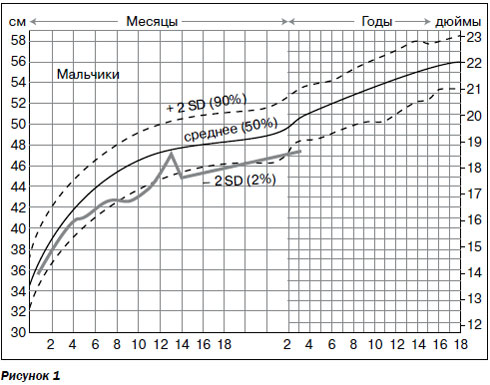
При осмотре у мальчика определяются микроцефалия (окружность головы 47,5 см), сходящееся альтернирующее косоглазие, динамическая и статическая атаксии, аутистическое поведение, афазия, частые стереотипии в виде облизывания рук, бруксизм, приступы гипервентиляции.
На основании данных окружности головы был составлен график прироста окружности, который показал прогрессирующий характер микроцефалии (рис. 1).
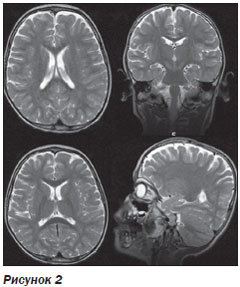
МРТ головного мозга выявило расширение субарахноидальных пространств в лобных отделах (рис. 2).
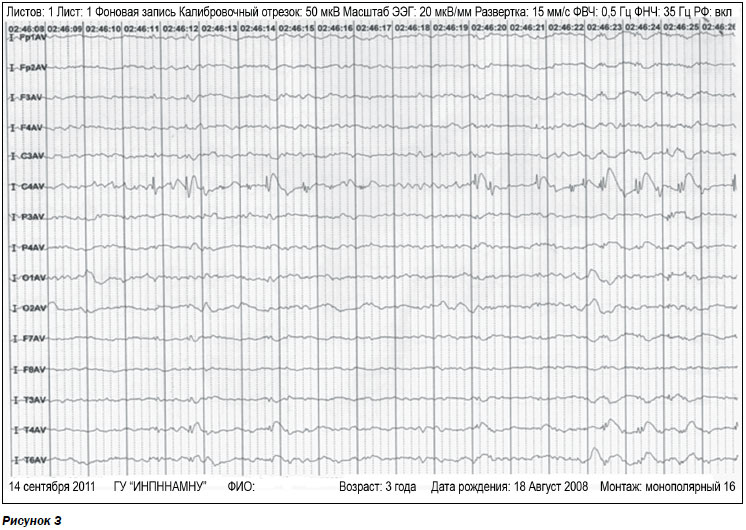
ЭЭГ показала наличие спайков и спайк-волновых комплексов в правой центральной области (рис. 3).
Также был проведен анализ свободных аминокислот крови, который оказался негативным в плане выявления болезней накопления.
Биохимический анализ крови (трансаминазы, триглицериды, мочевина, мочевая кислота, холестерол, креатинин, креатининкиназа, общий белок, альбумин) также оказался нормальным.
Кариотипирование выявило нормальный мужской 46,XY, SSCP-анализ кодирующей последовательности гена МеСР2, секвенсирование 3-го и 4-го экзонов гена МеСР2 выявили миссенс-мутацию в 4-м экзоне гена МеСР2:с.398 G->T, p.R133L, что подтвердило предположение о наличии у ребенка синдрома Ретта.
Дискуссия
Действительно, определение синдрома Ретта у мальчика является большой редкостью, но в данном случае диагностический поиск облегчался классическим течением заболевания. Типичное время начала заболевания (в 8 месяцев), утрата речи, навыков хождения, прогрессирующая микроцефалия, появление типичных стереотипий заставляли думать именно о синдроме Ретта. Нужно сказать, что нейрофизиологическое обследование (наличие центральных спайков и спайк-волновых комплексов), так же как и нейровизуализационное (атрофия лобных долей), необходимо для диагностики данного синдрома. Ну и самое главное — это определение мутации. В данном случае определение мутации в типичном месте, характерном для синдрома Ретта, ставит точку в диагностическом поиске.
1. Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood // Wien Med Wochenschr. — Sept. 1966. — 116(37). — 723-6. PMID 5300597.
2. Trappe R., Laccone F., Cobilanschi J. et al. MECP2 mutations in sporadic cases of Rett’s Disorder are almost exclusively of paternal origin // American journal of human genetics. — May 2001. — 68(5). — 1093-101.
3. Berridge C.W., Waterhouse B.D. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes // Brain Res. Rev. — 2003. — 42. — 33-84.
4. http://www.rettsearch.org/news_pubs.jsp
5. http://www.mychildwithoutlimits.org/?page=stages-of-rett-syndrome