
Газета «Новости медицины и фармации» Гастроэнтерология (419) 2012 (тематический номер)
Вернуться к номеру
Влияние альгинат-содержащего антирефлюксного препарата в форме суспензии на биодоступность омепразола
Авторы: P.W. Dettmar, F.C. Hampson, S.L. Little1, T. Baxter - Reckitt Benckiser Healthcare (UK) Ltd., Hull, UK; A. Jain, S. Choubey - Wellquest Research Pvt. Ltd., Wellspring Hospital, Mumbai, India
Версия для печати
Введение и цели. Омепразол применяют для лечения гастроэзофагеальной рефлюксной болезни (ГЭРБ). Этот препарат ингибирует секрецию кислоты, в то время как альгинат-содержащие антирефлюксные препараты действуют посредством образования «плота» низкой плотности с pH, близким к нейтральному, который покрывает содержимое желудка и физически препятствует развитию гастроэзофагеального рефлюкса. Данные в отношении возможных фармакокинетических взаимодействий между этими двумя препаратами ограничены, хотя их часто назначают одновременно для улучшения контроля симптомов у пациентов с ГЭРБ. Данное исследование было разработано для того, чтобы определить, влияет ли прием альгинат-содержащего препарата в форме суспензии на фармакокинетику омепразола.
Методы. Рандомизированное, перекрестное исследование 2 препаратов (омепразол и альгинат-содержащий препарат в виде суспензии) у 26 добровольцев. Каждый препарат назначали в течение 3 последовательных дней с 7-дневным периодом «вымывания» между фазами. Образцы крови для фармакокинетического анализа набирали в течение 24 часов после приема последней дозы омепразола.
Результаты и выводы. Прием Gaviscon Advance не влиял на кривую концентрации омепразола в плазме крови, то есть не усиливал влияние приема пищи на всасывание омепразола в форме MUPS таблеток. Таким образом, Gaviscon Advance — альгинат-содержащий препарат в форме суспензии, эффект которого реализуется физическим образом (путем образования плота-барьера) и не зависит от попадания в системный кровоток, не оказывает какого-либо влияния на фармакокинетику омепразола при его многократном приеме в течение 3-дневного периода времени.
Gaviscon® — это антирефлюксный препарат, который проявляет свой эффект посредством образования альгинатного плота, располагающегося («плавающего») на поверхности содержимого желудка и обеспечивающего таким образом физический барьер, предотвращающий рефлюкс кислоты в пищевод. Gaviscon® в форме суспензии оказывает эффект посредством взаимодействия альгината с кислотой желудка с образованием пленки с pH, близким к нейтральному. В ранее проведенных исследованиях было показано, что суспензия Gaviscon® образует прочный альгинатный плот-барьер in vitro и такой плот-барьер остается в верхней части желудка в течение 1–2 часов в отличие от антацидов или других препаратов, содержащих альгинат. В данном исследовании использовали альгинат-содержащий препарат в форме суспензии Gaviscon Advance® — он также образует прочный альгинатный плот-барьерin vitro, который сохраняется в желудке после эвакуации пищи. Механизм его действия не связан с абсорбцией в системный кровоток, потому лекарственные взаимодействия неизвестны.
Омепразол относится к антисекреторным препаратам, замещенным бензимидазолам, которые подавляют желудочную секрецию посредством специфического ингибирования H+/K+-аденозинтрифосфатной энзимной системы на секреторной поверхности желудочных париетальных клеток. Стабильность омепразола зависит от pH, и он быстро распадается в кислотной среде, но в щелочных условиях он обладает приемлемой стабильностью. Абсорбируется препарат быстро — пиковый уровень препарата в плазме достигается через 0,5–3,5 часа.
Пиковые концентрации омепразола в плазме и площадь под кривой «концентрация — время» (AUC) примерно пропорциональны при приеме в дозах до 20 мг и характеризуются высокой изменчивостью у разных людей. Абсолютная биодоступность (по сравнению с внутривенным введением) для доз 20–40 мг составляет 30–40 %, что в значительной степени связано с пресистемным метаболизмом. Период полураспада (T1/2) в плазме составляет 30–60 минут. Связывание с белками плазмы достигает 95 %.
Биодоступность омепразола несколько увеличивается при повторном приеме. Большая часть дозы (примерно 77 %) выводится с мочой в виде метаболитов. У пациентов с хронической патологией печени биодоступность повышается и плазменный T1/2 увеличивается до почти 3 часов. У пациентов с хронической патологией почек выведение омепразола подобно таковому у здоровых добровольцев, но отмечается некоторое повышение биодоступности.
Омепразол может пролонгировать элиминацию диазепама, варфарина и фенитоина. В отдельных случаях сообщалось о взаимодействии с другими препаратами, которые метаболизируются через систему цитохромов P450 (циклоспорин, дисульфирам, бензодиазепины). Омепразол может влиять на всасывание препаратов, для которых рН желудка является важным фактором, определяющим их биодоступность (например, кетоконазол, эфиры ампициллина и соли железа).
В Великобритании и других европейских странах омепразол и Gaviscon Advance® назначают в обычной практике и рекомендуют к применению в виде монотерапии или комбинации для симптоматического лечения ГЭРБ и сопровождающих ее симптомов — регургитации кислоты, изжоги и нарушения пищеварения. Поэтому желательно знать, существуют ли взаимодействия между этими двумя препаратами, которые могли бы влиять на фармакокинетику омепразола. Мы провели это исследование, чтобы проверить, влияет ли прием суспензии, содержащей альгинат, на фармакокинетический профиль омепразола у здоровых мужчин-добровольцев.
Материалы и методы
Дизайн исследования.Рандомизированное перекрестное исследование по изучению фармакокинетики омепразола в разных дозах (MUPS® [Multiple Unit Pellet System] таблетки Losec, 20 мг, компании Astra Zeneca, Великобритания) в сочетании с 10% суспензией альгината (Gаviscon Advance®, Reckitt Benckiser Healthcare, Великобритания) или без нее, которое было проведено в клинике Wellquest Research Pvt. Ltd., Wellspring Hospital, Мумбаи, в 2001–2002 гг. Пациенты были случайным образом распределены на две группы лечения. Лечение проводили в течение 3 дней (каждый период) — это достаточное время для того, чтобы омепразол достиг пиковой концентрации в плазме. Пациенты получали 1 препарат в первом периоде исследования, затем следовал стандартный период «вымывания» длительностью 7 дней и лишь потом — второй период. Исследование проводили открытым образом, так как не существует подходящего плацебо, которое позволило бы заменить суспензию, содержащую альгинат. Маловероятно, что открытый дизайн исследования мог бы повлиять на результаты, так как исследуемые показатели являлись объективными (плазменная концентрация) и образцы изучали специалисты, не знавшие о том, какое именно лечение принимал пациент.
Протокол исследования был рассмотрен и одобрен местным независимым этическим комитетом (IEC, Wellspring Hospital, Lower Parel, Мумбаи). Все пациенты были проинформированы о деталях исследования. До выполнения каких-либо процедур добровольцев устно ознакомили с формой о согласии, утвержденной IEC, которую каждый из них подписал.
Каждый волонтер получал в случайном порядке лечение А: омепразол магния 20,6 мг (эквивалент омепразола 20 мг) в таблетках перорально за 15 минут до завтрака в течение 3 дней с 240 мл питьевой воды; лечение В: омепразол магния 20,6 мг (эквивалент омепразола 20 мг) в таблетках перорально за 15 минут до завтрака плюс 10% раствор суспензии альгината (10 мл раствора, содержащие 1 мг альгината натрия и 0,2 г бикарбоната калия) перорально 4 раза в день (через 30 минут после приема пищи и перед сном) в течение 3 дней.
Образцы крови и анализы.Для оценки плазменной концентрации омепразола в течение 3 дней набирали повторные образцы крови. Для оценки показателей в последний день приема омепразола образцы крови набирали в период времени в 47,5–72 часа. Образцы крови (каждый по 5 мл) набирали в полипропиленовые трубки, содержавшие 5% раствор этилендиаминтетрауксусной кислоты. Образцы крови набирали за 30 минут до приема омепразола на 1-й и 3-й день и затем, после приема дозы, — на 3-й день через 0,5; 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 5,0; 6,0; 8,0; 12,0 и 24,0 часа после приема препарата. Образцы центрифугировали с целью отделить плазму и затем хранили при температуре –50 °C до проведения анализа.
Для определения омепразола в образцах крови применяли валидированный метод высокоэффективной жидкостной хроматографии (Welllquest Research Pvt. Ltd., Мумбаи, Индия). Плазму (500 μl) перемешивали с 100 μl 10 μl/мл пантопразола, встряхивали в 4 мл дихлорметана и центрифугировали; 3 мл экстракта дихлорметана испаряли до сухого состояния и затем восстанавливали в 250 μl жидкой фазы. Использовали колонки 4,6 ґ 150 мм с 5 μm Zorbax SB-C18, при t = 30 °C и 50 мМ дигидрофосфата натрия (рН 7,2)/ацетонитрил (75 : 25) в качестве подвижной фазы при скорости потока в 1 мл в минуту. Элюированный омепразол и пантопразол регистрировали с помощью ультрафиолетовых лучей с длиной волны 302 нм. Образцы плазмы исследуемых субъектов и стандартные смеси чередовали с образцами контроля качества, которые состояли из плазмы с омепразолом в концентрации от 15 до 1700 нг/мл; их обрабатывали таким же образом, как и образцы исследуемых. Все образцы сравнивали с кривой референтного стандарта — омепразола, приготовленного в концентрациях от 15 до 2000 нг/мл.
Нижний количественный предел определили на уровне 15,65 нг/мл, при котором точность составляла 7,87 % и достоверность — 99,18 %. В каждый отдельный день точность колебалась от 1,17 до 8,92 %, а достоверность — от 89,8 до 104,25 %. Точность между пробами в отдельные дни варьировала от 3,47 до 7,87 %, а достоверность — от 91,53 до 99,18 %. В образцы, предназначенные для контроля качества, добавляли известное количество омепразола в каждой партии изучаемых образцов, и, по возможности, образцы каждого из изучаемых субъектов анализировали с использованием одной и той же стандартной кривой. Образцы ниже количественного предела рассматривали как 0 при всех фармакокинетических и статистических оценках.
Фармакокинетический анализ.В образцах крови изучаемых субъектов определяли следующие фармакокинетические параметры, отражающие плазменную концентрацию омепразола: Tmax — время достижения максимальной концентрации препарата в плазме. Если максимальный показатель отмечали более чем в 1 временной точке, то Tmax определяли как значение в первой временной точке. Cmax — максимальная плазменная концентрация. AUC0-t — площадь под кривой «концентрация — время» от оценки до введения дозы и до последней измеряемой концентрации, рассчитанная с помощью метода трапеций. AUC0-a — площадь под кривой «концентрация — время» от оценки до приема первой дозы до бесконечности, которую рассчитывали как AUC0-t плюс отношение последней измеряемой концентрации к Kel. Kel — скорость выведения препарата из организма. T1/2 — период полувыведения препарата (время, необходимое для того, чтобы концентрация препарата в плазме снизилась вдвое по сравнению с первоначальной).
Статистические методы.Определяли средние арифметические (SD) показатели, стандартные отклонения и коэффициент вариации (CV) для неизмененных данных и геометрические показатели и CV для логарифмически преобразованных данных. Отмечали соотношения средних геометрических параметров для релевантных фармакокинетических показателей. Неизмененные и логарифмически преобразованные фармакокинетические параметры (Cmax, AUC0-t, AUC0-a) анализировали с использованием модели ANOVA. Для сравнения данных одного и того же субъекта (т.е. период, препарат и др.) применяли 5% уровень значимости, а для разных — 10%. При дисперсионном анализе рассчитывали наименьшие квадратичные значения, скорректированные различия между показателями и стандартные ошибки, связанные с этими различиями. Различия между субъектами для каждого фармакокинетического параметра, отражающие остаточную вариабельность после учета разницы между субъектами, фазы и терапии, выражали в виде общего коэффициента вариации (% CV) по результатам ANOVA с использованием логарифмически преобразованных данных.
90% ДИ для средних геометрических показателей фармакокинетических параметров (Cmax, AUC0-t, AUC0-a) рассчитывали на основании результатов ANOVA логарифмически преобразованных данных. 90% ДИ были основой для оценки эквивалентности изучаемых препаратов. В соответствии с критериями биоэквивалентности EuropeanAgencyfortheEvaluationofMedicinalProducts(EMEA), если соотношения и ДИ должны попадать в диапазон 80–125 %, то изучаемые методы лечения считают эквивалентными; в противном случае — не эквивалентными.
В исследовании с участием 16 здоровых мужчин-волонтеров среднее геометрическое AUC на 7-й день у лиц, получавших 20 мг омепразола один раз в день в течение 7 дней, составило 0,28 мг · ч/л и Cmax на 7-й день — 0,184 мг/л. На логарифмической шкале средний логарифмический показатель AUC был –1,273 (СО между субъектами — 0,693) и среднее логарифмическое Cmax –1,693 (СО между субъектами — 0,582). Однако нет подробных данных в отношении вариабельности этих параметров между субъектами.
Если средние логарифмически преобразованные показатели AUC для группы лечения (омепразол + альгинат) и контроля (омепразол) идентичны, то необходимо 11 человек; если это не так, но они предположительно варьируют в пределах 5 %, то необходимо 14 человек.
Для обеспечения 80% мощности, позволяющей показать эквивалентность лечения по такому показателю, как Cmax, требовался перекрестный дизайн с участием как минимум 29 человек при условии, что средние логарифмически преобразованные показатели Cmax для группы терапии и контроля одинаковы; если они не идентичны, но, возможно, находятся в пределах 5 %, то требуется 39 человек.
При использовании такого дизайна полный анализ 24 субъектов обеспечит достаточную статистическую мощность для того, чтобы показать эквивалентность в показателях AUC, и примерно 70% мощность, чтобы показать эквивалентность в Cmax если средняя логарифмически преобразованная Cmax для лечения и контроля одинаковы, и примерно 62% мощность, если допустить, что они находятся в пределах 5 %.
Таким образом, размер выборки в 24 человека рассматривался как достаточный для данного исследования. Для того чтобы удостовериться, что хотя бы 24 человека завершат исследование, было отобрано 26 субъектов для последующего скрининга и рандомизации.
Результаты
Оба периода исследования завершили 24 из 26 включенных субъектов. Все изучаемые были здоровыми мужчинами азиатского происхождения, которых отбирали в регионе Мумбаи (Индия). Полиморфизм CYP2C19 не определяли. Двое участников вышли из исследования после первого периода дозирования (по 1 из каждой группы) по личным причинам. Двое участников, завершивших исследование, сообщали о побочных эффектах: диарее на фоне лечения омепразолом в сочетании с альгинатом (расценили как возможно связанный с лечением) и повышении температуры тела на фоне лечения омепразолом (сочли не связанным с лечением).
Демографические и другие характеристики пациентов.Возраст, рост и масса тела исследуемых субъектов варьировали в пределах 18–28 лет, 160–175 см и 49–71 кг соответственно. Все не курили (за исключением 1 пациента, который сообщил, что жует табак) и не употребляли алкоголь. Все пациенты не принимали участие в клинических исследованиях и не сдавали кровь в объеме, превышающем 350 мл, в течение 3 месяцев до включения в данное исследование, а также не принимали медикаментозные препараты в течение 2 недель до того, как получить первую дозу изучаемого средства. Ни один из участников не имел клинически значимых событий в анамнезе и в семейном анамнезе, не страдал аллергией на животных, продукты питания или препараты. Не было выявлено клинически значимых нарушений во время общего и системного осмотра (сердечно-сосудистая, нервная, дыхательная и желудочно-кишечная системы) и обследования (электрокардиография и рентгенография органов грудной клетки).
Фармакокинетические результаты. Был проведен анализ данных 24 участников, его результаты представлены в табл. 1.

Геометрические показатели и соотношения выглядели следующим образом: Сmax — 555 для комбинации омепразол/альгинат и 558 — для омепразола (соотношение 99,55 %, 90% ДИ 82,75–119,75 %; AUC0-t — 2050 для комбинации омепразол/альгинат и 2094 — для омепразола (соотношение 97,90 %, 90% ДИ 87,83–109,12 %); AUC0-a — 2247 для комбинации омепразол/альгинат и 2231 — для омепразола (соотношение 100,74 %, 90% ДИ 90,05–112,70 %). Средние значения Tmax, Kel и T1/2 также были подобными на фоне приема обоих вариантов лечения. Средние концентрации омепразола в плазме в течение дня после введения последней дозы представлены на рис. 1. 90% доверительные интервалы для средних геометрических показателей для Cmax, AUC0-t и AUC0-a укладываются в интервале 80–125 % и, таким образом, соответствуют критериям биоэквивалентности EMEA.
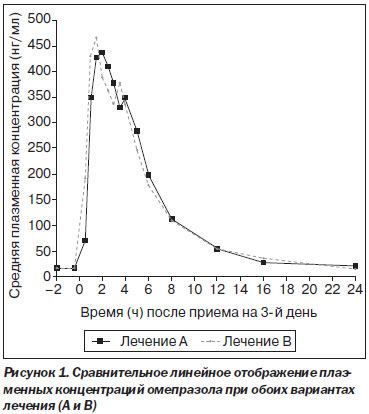
Обсуждение
Лекарственная форма омепразола оказывает значительное влияние на его фармакокинетику, что отчасти связано с нестабильностью препарата в кислотной среде. Твердые лекарственные формы в основном представлены покрытыми кишечнорастворимой оболочкой гранулами, и хотя они могут иметь такой же показатель AUC, как и буферная жидкая доза, максимальная концентрация в плазме ниже и достигается позже. Было показано, что твердая лекарственная форма омепразола, которую применяли в этом исследовании (MUPS таблетки), биоэквивалентна после 1 и 6 дней приема более старой, широко применяемой форме покрытых кишечнорастворимой оболочкой гранул в твердых желатиновых капсулах. Это означает, что основные фармакокинетические параметры омепразола, которые были получены в этой работе (AUC, Cmax, T1/2, Kel), можно сравнивать с таковыми, полученными в работах с применением капсул и в немногочисленных исследованиях с применением MUPS таблеток. Очевидно, что фармакокинетические результаты делятся на две группы в зависимости от этнического происхождения субъектов (кавказец или азиат). Как известно, это связано с основным ферментом, участвующим в метаболизме омепразола, — CYP2C19. У некоторых людей CYP2C19 является дефицитным, в результате чего концентрация в плазме и AUC в четыре или пять раз выше, чем у лиц с этой изоформой. Дефицит также приводит к трехкратному повышению T1/2 у лиц со сниженным метаболизмом. Известно, что генетический вариант, при котором имеется недостаточность CYP2C19, гораздо более распространен в Азии, чем среди кавказцев, хотя сообщалось о его наличии и у японцев, китайцев и корейцев. Фармакокинетические параметры, полученные в этом исследовании, сопоставимы с результатами предыдущих исследований с участием индийцев и австралийцев. Эти данные не соответствуют результатам некоторых исследований c применением кишечнорастворимых капсул, проведенных в Европе и Соединенных Штатах: так, при повторном приеме AUC составляла от 1/4 до 1/5 от показателя, полученного в данном исследовании. В этих работах Tmax находился в пределах 1,25–1,6 часа, что существенно меньше, чем 2,2 часа в данной работе. В ходе двух проведенных в Европе исследований биодоступности MUPS таблеток были получены типичные фармакокинетические параметры для кавказцев.
Существуют противоречивые данные о влиянии антацидов на биодоступность омепразола в кишечнорастворимых гранулах. В двух европейских исследованиях не было выявлено влияния жидких антацидов, содержащих алюминия и магния гидроксид, на фармакокинетические показатели после одной дозы омепразола, в то время как в японском исследовании наблюдали значительное снижение AUC при приеме омепразола в кишечнорастворимых таблетках в сочетании с гранулами Maalox, но не с суспензией Maalox. В нашем исследовании не было отмечено отличий в биодоступности, когда Gaviscon Advance® применяли с омепразолом в MUPS таблетках, но это может быть связано с очень слабыми кислотонейтрализующими свойствами Gaviscon Advance® и отсутствием в его составе вышеупомянутых антацидов — гидроксидов алюминия и магния. Gaviscon Advance® имеет форму суспензии и, попадая в желудок, формирует плавающую желатиновую массу, которая задерживается в желудке. Влияние пищи на биодоступность омепразола в кишечнорастворимых гранулах состоит в замедлении всасывания препарата, при этом общая доступность не снижается. В данном исследовании омепразол принимали за 15 мин до завтрака, Gaviscon Advance® — через 30 минут после. Полученные результаты свидетельствуют о том, что прием Gaviscon Advance® не влиял на концентрацию омепразола в плазме крови, то есть не усиливал влияния пищи на всасывание омепразола в форме MUPS таблеток.
Так как 90% доверительные интервалы для Cmax, AUC0-t и AUC0-a укладываются в диапазон 80–125 %, можно сделать вывод, что при сочетанном применении омепразола и альгинат-содержащего препарата в форме суспензии Gaviscon Advance®, эффект которого реализуется физическим образом (путем образования плота-барьера) и не зависит от попадания в системный кровоток, альгинат-содержащий препарат не оказывает какого-либо влияния на фармакокинетику омепразола при его многократном приеме в течение 3-дневного периода.
Оригинал статьи опубликован в
Indian. J. Med. Res., 123, April 2006, р. 517-524
Перевод К. Кремца
1. Washington N., Washington C., Wilson C.G., Davis S.S. The effect of inclusion of aluminium hydroxide in alginatecontaining raft-forming antacids // Int. J. Pharmaceut. — 1986. — 28. — 139-43.
2. Bennett C.E., Hardy J.G., Wilson C.G. The influence of posture on the gastric emptying of antacids // Int. J. Pharmaceut. — 1984. — 21. — 341-7.
3. May H.A., Wilson C.G., Hardy J.G. Monitoring radiolabelled antacid preparations in the stomach // Int. J. Pharmaceut. — 1984. — 19. — 169-76.
4. Washington N., Washington C., Wilson C.G. Gastric distribution and residence time of two anti-reflux formulations // Int. J. Pharmaceut. — 1987. — 39. — 163-71.
5. Burkitt V., Dettmar P.W., Jolliffe I.G., Sykes J. Evaluation of the barrier properties of UK alginate suspensions // Gut. — 2000. — 47 (Suppl. III). — A57.
6. Marciani L., Little S.L., Coleman N., Young P., Taylor D., Snee J. et al. In vivo visualisation of Gaviscon alginate rafts using echo-planar magnetic resonance imaging // Gut. — 2001. — 48 (Suppl. I). — A36.
7. Fellenius E., Berglindh T., Sachs G., Olbe L., Elander B., Sjostrand S.E. et al. Substituted benzimidazoles inhibit gastric acid secretion by blocking (H+/K+) ATPase // Nature. — 1981. — 290. — 159-61.
8. Regardh C.-G., Gabrielsson M., Hoffmann K.-J., Lofberg I., Skanberg I. Pharmacokinetics and metabolism of omeprazole in animals and man — an overview // Scand. J. Gastroenterol. — 1985. — 20 (Suppl. 108). — 79-94.
9. Andersson T., Olsson R., Regardh C.G., Skanberg I. Pharmacokinetics of [14C] omeprazole in patients with liver cirrhosis // Clin. Pharmacokinet. — 1993. — 24. — 71-8.
10. Naesdal J., Andersson T., Bodemar G., Larsson R., Regardh C.-G., Skanberg I. et al. Pharmacokinetics of [14C] omeprazole in patients with impaired renal function // Clin. Pharmacol. Ther. — 1986. — 40. — 344-51.
11. Andersson T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors. Focus on omeprazole, lansoprazole and pantoprazole // Clin. Pharmacokinet. — 1996. — 31. — 9-28.
12. Note for guidance on the investigation of bioavailability and bioequivalence. CPMP/EWP/QWP/140/98. EMEA, 2001.
13. Hartmann M., Theiss U., Huber R., Luhman R., Bliesath H., Wurst W. et al. Twenty four hour intragastric pH profiles and pharmacokinetics following single and repeated oral administration of the proton pump inhibitor pantoprazole in comparison to omeprazole // Aliment. Pharmacol. Ther. — 1996. — 10. — 359-66.
14. Nikiforova O.V., Chistyakov V.V., Shikh E.V., Arzamastsev A.P. Pharmacokinetics and bioavailability of gastrozole // Pharmaceut. Chem. J. — 1999. — 33. — 173-6.
15. Farinha A., Bica A., Pais J.P., Toscano M.C., Tavares P. Bioequivalence evaluation of two omeprazole enteric-coated formulations in humans // Eur. J. Pharm. Sci. — 1999. — 7. — 311-5.
16. Pilbrant A., Cederberg C. Development of an oral formulation of omeprazole // Scand. J. Gastroenterol. — 1985. — 20 (Suppl. 108). — 113-20.
17. Anderberg E.K., Cullberg M., Langstrom G., Naesdal J., Rosen E. Bioequivalence between omeprazole MUPS® tablets and omeprazole capsules // Gastroenterology. — 1998. — 114. — A56.
18. Ishizaki T., Sohn D.R., Kobayashi K., Chiba K., Lee K.H., Shin S.G. et al. Interethnic differences in omeprazole metabolism in the two S-mephenytion hydroxylation phenotypes studied in Caucasians and Orientals // Ther. Drug Monit. — 1994. — 16. — 214-5.
19. Sane R.T., Ghadge J.K., Jani A.B., Vaidya A.J., Kotwal S.S. Comparative evaluation of pharmacokinetic and bioequivalence study of omeprazole capsules in healthy Indian men // Indian Drugs. — 1992. — 29. — 445-6.
20. Pritchard P.J., Yeomans N.D., Mihaly G.W., Jones D.J., Buckle P.J., Smallwood R.A. et al. Omeprazole: a study of its inhibition of gastric pH and oral pharmacokinetics after morning or evening dosage // Gastroenterology. — 1985. — 88. — 64-9.
21. Duvauchelle T., Millerioux L., Gualano V., Evene E., Alcaide A. Comparative bioavailability study of two oral omeprazole formulations after single and repeated administrations in healthy volunteers // Clin. Drug Investig. — 1998. — 16. — 141-9.
22. Sharma V.K., Peyton B., Spears T., Raufman J.P., Howden C.W. Oral pharmacokinetics of omeprazole and lansoprazole after single and repeated doses as intact capsules or as suspensions in sodium bicarbonate // Aliment. Pharmacol. Ther. — 2000. — 14. — 887-92.
23. Geus W.P., Mathot R.A.A., Mulder P.G.H., Lamers C.B.H.W. Pharmacodynamics and kinetics of omeprazole MUPS 20 mg and pantoprazole 40 mg during repeated oral administration in Helicobacter pylori-negative subjects // Aliment. Pharmacol. Ther. — 2000. — 14. — 1057-64.
24. Schaltenbrand R., Huber R., Cotoraci C.A., Mascher H., Potthast H., Hole U. Bioequivalence between omeprazole MUPS 20 mg as tablet and omeprazole MUPS 20 mg as tablet encapsulated in a hard gelatine capsule // Int. J. Clin. Pharmacol. Ther. — 2001. — 39. — 453-9.
25. Tuynman H.A.R.E., Festen H.P.M., Meuwissen S.G.M. Pharmacokinetic studies with omeprazole: the effect of antacids on the bioavailability of omeprazole // Neth. J. Med. — 1986. — 29. — 148.
26. Iwao K., Saitoh H., Takeda K., Azuumi Y., Takada M. Decreased plasma levels of omeprazole after coadministration with magnesium-aluminium hydroxide dry suspension granules // Yakugaku Zasshi. — 1999. — 119. — 221-8.