Введение
Наследственная оптическая нейропатия Лебера (LHON) является заболеванием, передающимся от матери к потомству, в основе которого лежит митохондриальная дегенерация ганглионарных клеток сетчатки и их аксонов, что приводит клинически к безболезненной острой или подострой двусторонней потере центрального зрения. Данное заболевание впервые было описано в 1871 году немецким офтальмологом Теодором Лебером [1]. В последующем, в 1964 году, было опубликовано описание сочетания LHON и рассеянного склероза у одного пациента [6]. Спустя практически 30 лет (в 1992 году) были опубликованы уже несколько случаев диагностированного сочетания LHON и рассеянного склероза группой авторов во главе с профессором Anita Harding, в честь которой данное сочетание в последующем и стали называть Harding-синдромом [2]. При последующем детальном исследовании LHON и рассеянного склероза было отмечено, что в обоих случаях наблюдается митохондриальная патология, которая занимает ведущую позицию в патогенезе. Такая схожесть в митохондриальных нарушениях данных заболеваний, вероятно, и привела к тому, что после активного внедрения магнитно-резонансной томографии (МРТ) при LHON были выявлены очаговые изменения в центральной нервной системе (ЦНС), которые не имеют специфических отличий от очагов демиелинизации при рассеянном склерозе [7, 8]. Морфологических отличий очаговых изменений и при LHON, и при рассеянном склерозе к настоящему времени не выявлено, и, более того, были отмечены схожие атрофические процессы [9]. В связи с этим при диагностике у одного пациента и LHON, и рассеянного склероза возникает ряд вопросов в отношении этиологии очаговых изменений по данным МРТ, что имеет большое значение в определении тактики ведения. В подтверждение неоднозначности современной трактовки очаговых изменений при сочетании LHON с рассеянным склерозом (Harding-синдром) приводим клинический случай.
Клинический случай
На базе Межокружного отделения рассеянного склероза при ГБУЗ «ГКБ № 24 ДЗМ» совместно с отделением лучевой диагностики ФГБНУ «Научный центр неврологии» наблюдается молодая пациентка 1997 года рождения с отягощенной наследственностью по материнской линии в отношении болезни Лебера и сахарного диабета.
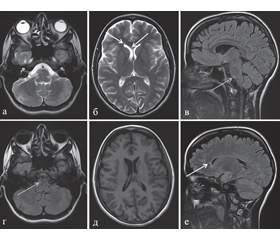
Дебют заболевания развился в сентябре 2011 года в виде снижения зрения на правый глаз, не сопровождающегося болевым синдромом, и пареза правого лицевого нерва, а через 2 недели присоединилось двоение. На МРТ головного мозга от 03.11.2011 было выявлено три очага повышенной интенсивности МР-сигнала в режимах Т2 и Т2 FLAIR: в задних нижних правых отделах варолиева моста, правых отделах колена мозолистого тела, глубоких отделах белого вещества лобно-теменной области правого полушария большого мозга (рис. 1). После введения контрастного вещества патологического накопления последнего не обнаружено. При проведении МРТ спинного мозга очаговых изменений выявлено не было. Диагностирован оптический неврит справа без болевого синдрома. По месту жительства была проведена гормональная пульс-терапия с частичным регрессом неврологической симптоматики, и в последующем осуществлялся клинико-МРТ-мониторинг.
/100-1.jpg)
Следующее ухудшение состояния пациентка отметила в октябре 2012 года в виде повторного эпизода снижения зрения на правый глаз без болевого синдрома. При проведении МРТ головного мозга по сравнению с предыдущим исследованием отмечается появление двух новых очагов: в центральных отделах колена мозолистого тела и передних отделах левого полушария мозжечка, а очаг в варолиевом мосту уменьшился в размерах (рис. 2). После введения контрастного вещества патологического накопления последнего не выявлено. При исследовании толщины зрительных нервов по данным МРТ головного мозга в режиме Т2 FatSat (с подавлением сигнала от жировой ретробульбарной клетчатки) было обнаружено уменьшение толщины обоих зрительных нервов до 0,2 см (при норме 0,3–0,4 см) (рис. 3). Проведена генетическая консультация на предмет болезни Лебера — выявлена мутация 3460G/A. При исследовании ликвора был обнаружен интратекальный синтез олигоклональных IgG. Выставлен диагноз: болезнь Лебера + ремиттирующий рассеянный склероз (Harding-синдром). Назначена терапия препаратом интерферон бета-1а, 44 мкг, подкожно, 3 раза в неделю.
/101-1.jpg)
На фоне назначенной терапии проводилось клинико-МРТ-наблюдение, в ходе которого какой-либо динамики не отмечалось до марта 2014 года. В марте 2014 года на фоне полного клинического благополучия на МРТ спинного мозга с контрастным усилением выявлен интрамедуллярно на уровне нижних отделов тела Th6-позвонка очаг с нечеткими контурами (рис. 4). После введения контрастного вещества патологического накопления последнего не обнаружено. По данным МРТ головного мозга отрицательная динамика не выявлена. Терапия препаратом интерферон бета-1а 44 мкг продолжена. При последующем клинико-МРТ-мониторинге какой-либо динамики обнаружено не было до осени 2015 года, когда на повторной МРТ головного мозга с контрастным усилением и по сравнению с предыдущими данными было определено появление нового очага в правых отделах варолиева моста (рис. 5). После введения контрастного вещества патологического накопления последнего не выявлено. Остальные очаги имеют прежние характеристики и несколько уменьшились в размерах.
/102-1.jpg)
Таким образом, представлен клинический случай сочетания LHON и рассеянного склероза у молодой пациентки с отягощенной наследственностью в отношении носительства мутации болезни Лебера по материнской линии. Диагноз болезни Лебера подтвержден генетически — обнаружена мутация 3460G/A. Диагноз «рассеянный склероз» подтвержден согласно критериям Макдонольда от 2010 года. Данной пациентке в соответствии со стандартами лечения рассеянного склероза назначена терапия препаратом интеферон бета-1а 44 мкг, на фоне чего отмечается появление новых очагов, что, с одной стороны, можно расценивать как субклиническую неэффективность проводимой патогенетической терапии рассеянного склероза, а с другой — как естественное течение болезни Лебера, для которой, как известно, также может быть характерно образование очагов в ЦНС.
Обсуждение
Исходя из проведенного анализа опубликованных к настоящему времени данных, становится очевидным, что дебют заболевания LHON в среднем приходится на конец подросткового возрастного периода. Выделяют и некоторые гендерные особенности LHON и Harding-ма: так, изолированный LHON чаще встречается у мужчин (до 77 % случаев), но именно сочетание c рассеянным склерозом наблюдается чаще среди женщин (примерно до 2/3 случаев) [16]. В связи с этим в настоящее время преобладает гипотеза о том, что LHON у женщин может быть фактором риска развития либо радиологически изолированного синдрома, либо рассеянного склероза [17].
Ряд проведенных научных исследований показал, что в основе патогенеза LHON лежат точечные мутации митохондриальной ДНК. Так, например, согласно данным, опубликованным в 1988 году D.C. Wallace с соавторами, атрофия зрительных нервов при LHON связана с заменой 11778-го нуклеотида митохондриального гена, который кодирует 4-ю субъединицу комплекса I дыхательной цепи. В последующем проводились более углубленные исследования и были обнаружены и другие мутации, ведущие к этой болезни. Было также отмечено, что в большинстве случаев мутации затрагивают митохондриальные гены, кодирующие белки, которые участвуют в процессе переноса электронов в дыхательной цепи, что приводит к нарушениям фосфорилирующей функции митохондрий. К настоящему времени известно, что в 95 % случаев в митохондриальной ДНК выявляются три мутации при LHON: в 11778, 3460 и 14484-м положениях [2–5]. Считается также, что именно митохондриальные дефекты при LHON запускают и аутоиммунный воспалительный процесс. Целый ряд современных научных исследований демонстрирует, что именно наличие митохондриальной дисфункции играет ключевую роль в формировании очагового поражения, аксонального повреждения и нейродегенерации при рассеянном склерозе [11–15]. Именно различия в митохондриальной ДНК и ядерной кодировке митохондриальных генов при LHON могут повлиять на генетическую предрасположенность к рассеянному склерозу [10]. Но, несмотря на это, к настоящему времени истинный генез очаговых изменений в ЦНС при LHON/Harding-синдроме остается дискутабельным, и в связи с этим вопрос касательно алгоритма лечения данной категории больных также остается открытым.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Man P. Leber hereditary optic neuropathy // Journal of Medical Genetics. — 2002. — 39(3). — 162-169. — doi: 10.1136/jmg.39.3.162.
2. Harding A., Sweeney M., Miller D. et al. Occurrence of a multiple sclerosis — like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation // Brain. — 1992. — 115(4). — 979-989. — doi: 10.1093/brain/115.4.979.
3. Palace J. Multiple sclerosis associated with Leber’s Hereditary Optic Neuropathy // Journal of the Neurological Sciences. — 2009. — 286(1–2). — 24-27. — doi: 10.1016/j.jns.2009.09.009.
4. Giordano C., Carelli V. Reply: Mitochondrial DNA copy number differentiates the Leber’s hereditary optic neuropathy affected individuals from the unaffected mutation carriers // Brain. — 2015. — 139(1). — 2. — doi: 10.1093/brain/awv217.
5. Riordan-Eva P., Sanders M., Govan G., Sweeney M., Costa J., Harding A. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation // Brain. — 1995. — 118(2). — 319-337. — doi: 10.1093/brain/118.2.319.
6. Lees F., Macdonald A., Turner J. Leber’s disease with symptoms resembling disseminated sclerosis // Journal of Neurology, Neurosurgery & Psychiatry. — 1964. — 27(5). — 415-421. — doi: 10.1136/jnnp.27.5.415.
7. Jansen P., van der Knaap M., de Coo I. Leber’s hereditary optic neuropathy with the 11 778 mtDNA mutation and white matter disease resembling multiple sclerosis: clinical, MRI and MRS findings // Journal of the Neurological Sciences. — 1996. — 135(2). — 176-180. — doi: 10.1016/0022-510x(95)00287-c.
8. Kovacs G. Neuropathology of white matter disease in Leber’s hereditary optic neuropathy // Brain. — 2004. — 128(1). — 35-41. — doi: 10.1093/brain/awh310.
9. Matthews L., Enzinger C., Fazekas F. et al. MRI in Leber’s hereditary optic neuropathy: the relationship to multiple sclerosis // Journal of Neurology, Neurosurgery & Psychiatry. — 2014. — 86(5). — 537-542. — doi: 10.1136/jnnp-2014-308186.
10. Ban M., Elson J., Walton A. et al. Investigation of the Role of Mitochondrial DNA in Multiple Sclerosis Susceptibility // PLoS One. — 2008. — 3(8). — 2891. — doi: 10.1371/journal.pone.0002891.
11. Mahad D., Ziabreva I., Lassmann H., Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions // Brain. — 2008. — 131(7). — 1722-1735. — doi:10.1093/brain/awn105.
12. Lu F., Selak M., O’Connor J. et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis // Journal of the Neurological Sciences. — 2000. — 177(2). — 95-103. — doi: 10.1016/s0022-510x(00)00343-9.
13. Dutta R., McDonough J., Yin X. et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients // Annals of Neurology. — 2006. — 59(3). — 478-489. — doi: 10.1002/ana.20736.
14. Mahad D., Lassmann H., Turnbull D. Review: Mitochondria and disease progression in multiple sclerosis // Neuropathology and Applied Neurobiology. — 2008. — 34(6). — 577-589. — doi: 10.1111/j.1365-2990.2008.00987.x.
15. Campbell G., Ziabreva I., Reeve A. et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis // Annals of Neurology. — 2010. — 69(3). — 481-492. — doi: 10.1002/ana.22109.
16. Pfeffer G., Burke A., Yu-Wai-Man P., Compston D., Chinnery P. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations // Neurology. — 2013. — 81(24). — 2073-2081. — doi: 10.1212/01.wnl.0000437308.22603.43.
17. Брюхов В.В., Попова Е.В., Кротенкова М.В., Бойко А.Н. Радиологически изолированный синдром (МРТ-критерии и тактика ведения больного) // Журнал неврологии и психиатрии им. C.C. Корсакова. — 2016. — 116. — 10(2). — 52-57.