Introduction
Von Willebrand’s disease (VWD), or von Willebrand’s Syndrome, is a bleeding syndrome characterized by low plasma levels of von Willebrand factor (VWF). VWD is the commonest inherited human bleeding disorder. Partial quantitative deficiency of serum VWF is responsible for the majority of VWD cases [1]. The effect of VWF deficiency on orthopedic operations is not well documented in the current literature [2]. VWD has been described following vascular surgery, coronary artery bypass grafting, renal and liver transplants, and abdominal surgeries.
VWD may cause persistent bleeding during the operative and postoperative periods [3]. In the majority of cases, VWD occurs as a single episode, but frequent relapses with chronicity can be seen in a small number of cases [4]. Patients should therefore receive long-term follow-up [5].
We reported a case of a 22-year-old man with VWD operated with intramedullary nailing due to tibial shaft fracture. The patient had no previous history of surgery, and was unaware of his VWD. The purpose of this study is to report a rare complication of an orthopedic surgical procedure with postsurgical bleeding mimicking tibialis anterior arterial perforation. Orthopedic surgeons must be alert to the possibility of VWD due to postsurgical difficulties and persistent bleeding.
Report of the Case
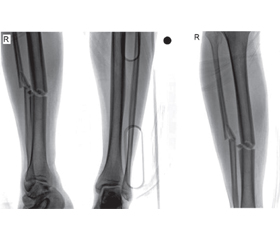
A 22-year-old male patient with unilateral/isolated tibial shaft fracture was admitted to our emergency department following a traffic accident. His medical records indicated no systemic disease. His preoperative complete blood count was normal (hemoglobin, white blood cell count, platelet count, coagulation parameters, and biochemical indexes were all normal). After one night, the patient was prepared for tibial nailing of tibial shaft fracture classified as AO/OTA 42A2 (fig. 1). The patient had no history of hemophilia. During surgery, abnormal bleeding was observed while the proximal screws were being inserted. Once the bleeding had been brought under control, the patient was sent to postoperative care and the orthopedic ward. Antibiotics (second generation cephalosporins) were given for infection prophylaxis (one dose pre-operatively and for 24 h intravenously). The patient felt well at the first postoperative clinical assessment. However, a number of purpuric rashes in the form of ecchymosis were observed in the perioperative region. On the postoperative second day, hemoglobin levels decreased by approximately one unit in 24 h. The patient exhibited subfebrile fever (37.6 °C), but there were no clinical symptoms. On the postoperative third day, palor was observed on the skin and mucosal surfaces, together with petechiae on the fractured limb, with mild swelling. There was no bleeding in the surgical area, with only ecchymoses being seen following the venous vascular traces. The patient’s general health was stable at evaluation in the postoperative period. No infection, septicemia, pneumonia, or urinary tract infection were observed. The patient was assessed by cardiovascular surgery (CVS) specialists and computed tomography angiography (CTA) was performed. Unclear perforation imaging was seen at CTA (fig. 2), but this was not confirmed by the CVS specialist. Complete blood count revealed progression to severe blood loss (hemoglobin from 13.37 to 8.40 g/dl, hematocrit (Ht) from 41 to 26 %, and platelet count from 121.000/mm3 to 50.000/mm3). Coagulation and bleeding time tests were normal (table 1, 2). During investigation of the family history, the patient’s mother described a history of bleeding diagnosed before the age of 22 at the birth of her son. She added that she had forgotten to provide this information before the operation.
/48.jpg)
In the light of the family history, a detailed hematological laboratory study was conducted. All laboratory values were evaluated due to the patient’s own and family history and clinical findings, and von Willebrand type 2 disease was eventually diagnosed.
After diagnosis, fresh frozen plasma (FFP) was administered, and the patient was transferred to the hematology department. Hematological and biochemical profiles were reported as optimized at follow-up (Htc: 30.5 %, PLT: 201.500/μl).
During that period, clinical status improved very rapidly, with no complications such as compartment syndrome. The patient was able to walk with crutches, but was walking independently by the end of the second month. Fusion mass was seen in the second month. The patient has been followed-up for the last x months.
Discussion
VWF is a large hemostatic protein produced by endothelial cells and megakaryocytes. It is a complex multimeric protein with two functions, forming a bridge between platelets and the damaged vascular area, and binding to and stabilizing factor VIII. VWF is important for platelet adhesion to the exposed collagen in the damaged vascular endothelial areas, and also contributes to the fibrin clotting by serving as a shuttle for coagulation protein factor VIII (FVIII), preventing its degradation by plasma proteases. The three functional aspects of VWF can be described as platelet binding, collagen binding, and FVIII binding. The D1 and D2 domains make up the VWF propeptide and result in the production of mature VWF. The D′ and D3 domain bind factor VIII (FVIII), the A1 domain binds platelets (PLT) and collagen (COL), and the A3 domain also binds collagen [6].
VWD is a genetic and multifactorial disease exhibiting autosomal dominant inheritance. Pregnancy, estrogen, progesterone, glucocorticoids, adrenalin-releasing states (such as stress and fear), intense physical exercise, hyperthyroidism, inflammatory disease, vasculitis, diabetes mellitus, and liver-kidney diseases all increase VWF levels, while hypothyroidism and valproic acid reduce them. VWF levels are also 25 % lower in the population with the O Rh blood group.
The prevalence of VWD is approximately 1 %, and it constitutes 10 % of bleeding symptoms. VWF is synthesized in megakaryocytes and stored in alpha granules. Adhesion and aggregation are performed by VWF. Bleeding may occur in case of functional insufficiency or low VWF levels. Clinical symptoms of VWD include such clinical changes as a greater disposition to bleeding, nasal-gingival bleeding, postpartum bleeding, post-incision bleeding, gastrointestinal bleeding, prolonged bleeding time, hemorrhage after surgery, menorrhagia, and easy bruising. Nasal bleeding is also common. Laboratory findings include prolongation of bleeding time, low VWF levels, low VWF-ristocetin co-factor activity, decreased ristocetin-platelet aggregation, and PFA 100 positivity. VWF levels, RCoF levels, RCoF test results, collagen binding activity (CBA), and FVIII activity can also be measured. Multimeric analysis and genetic tests can also be conducted for the diagnosis of VWD.
There are thre types of VWD. Type 1 involves a partial lack of VWF (75–80 % of all types; VWF levels greater than 30%), while type 2 occurs when VWF exhibits structural (quantitative) abnormality. Type 2 (VWF levels of 1–30 %) also consists of four subtypes — 2A (15–20 %), 2B (5 %), 2M (very rare), and 2N (autosomal recessive inheritance). Type 3 is defined as complete absence of VWF (less than 1 %, with autosomal recessive inheritance). If the patient’s VWF is lower than 30 IU/dl, significant bleeding may occur. However, individuals with VWF levels of 30–50 IU/dl cannot be diagnosed unless bleeding occurs [7].
VWD involves quantitative VWF defects, either partial (type 1 with VWF levels < 50 IU/dL) or almost total (type 3 with undetectable VWF levels), and also qualitative VWF defects (type 2 variants with discrepant antigenic and functional VWF levels) [8]. Diagnostic tests including activated partial thromboplastin time, bleeding time, factor VIII:C Ristocetin cofactor and VWF antigen, can be performed for diagnosis of VWD when this is suspected. Ristocetin-induced platelet adhesion, the multimeric structure and collagen binding test, and genetic analysis are of assistance with diagnosis of VWD. However, suspicion represents the first pre-requisite for diagnosis [8]. In the guidelines, patients with marked reductions in plasma VWF levels (< 30 IU/dL) usually have significant bleeding phenotypes and should be classified as type 1 VWD. The second type, involving cases with intermediate reduced plasma VWF:Ag levels (in the range of 30–50 IU/dL), patients exhibit variable bleeding phenotypes and often do not have VWF gene sequence variations of the undefined type [1].
Definitive diagnosis is based on the following diagnostic parameters: 1) mucocutaneous bleeding, 2) low levels of VWF, and 3) history/family history. Treatment should consist of local hemostatics, anti-fibrinolytic, hormonal therapy, and replacement coagulation products. Local hemostasis is performed using local physical pressure, sponge gels, and surgicel. Tranexamic acid (Transamin) is used as an anti-fibrinolytic at doses of 250–500 mg. Analogue estrogens and LHRH may also be used. Fresh frozen plasma (FFP), cryoprecipitates, FVIII/VWF concentrations, and platelet transfusions are employed in replacement therapy [9].
One previous study evaluated VWF levels in 1218 participants. The ristocetin co-factor activity of VWF was measured essentially using the method described by Macfarlane et al. [10]. The Newman Keuls test also demonstrates lower levels of VWF. The male/female ratio in this study group was 1.01. In terms of clinical symptoms, epistaxis accounted for 38 % of cases, bleeding after tooth extraction for 20 %, menorrhagia for 18 %, postpartum hemorrhage for 8 %, prolonged bleeding after superficial cuts for 4%, tonsillectomy/adenoidectomy for 4.5 %, post-surgical bleeding for 3.5 %, easy bruising for 2 %, and other causes for 1 %. The frequency of bleeders in the general population is unknown. Miller et al. classified as many as 23 % of normal subjects as symptomatic, a figure that might undermine the discriminant ability of this parameter [11]. The prevalence of VWF in the population is thus estimated at between 0.57 and 1.15 % heterozygotic and autosomal dominant. Type 1 exhibits a mutant gene carrier rate of approximately 2 %, with an estimated prevalence in the general population of 25/1.000.000. Considering the frequency of VWD suggested by this study, we think that a VWF assay (ristocetin cofactor activity) should be included among the first level screening tests for evaluating subjects with mild bleeding diathesis even in the presence of normal routine screening tests. An epidemiological investigation among schoolchildren in the Veneto region of Northern Italy by Rodeghiero et al. [12], in which 1218 of the 1281 children participating were aged between 11 and 14. Blood samples were collected from all individuals for determination of blood groups and VWF levels (measured as ristocetin cofactor and expressed in IU/dL after calibration of the internal pool against an international standard). A questionnaire was then administered concerning hemorrhagic symptoms in family members over the last three generations. The authors suggested that the prevalence of VWD may be much higher than previously reported and that a different screening approach might be of use for patients with a mild bleeding diathesis ranging from 7 (0.57 %) to 14 (1.15 %) [13, 14].
In the largest series of fractures in individuals with hemophilia (PWH), Caviglia et al. established a higher incidence of lower limb (LL) fractures in the first period analyzed (1986–1990). The LL/upper limb ratio changed as UL fractures became more frequent. The authors emphasized that the advent of new and accessible treatments had reduced the development of orthopedic complications and resulted in the improved quality of life in PWH [15].
In another study, thrombotic thrombocytopenic purpura (TTP), another blood disorder, was diagnosed with bleeding complications following othopedic surgery [16]. Those authors recommended a treatment modality of urgent plasma exchange, plasmapheresis, initial treatment with plasma infusion and glucocorticoids, plasma infusion, γ-globulin or glucocorticoid administration, and antiplatelet drugs (dipyridamole and aspirin). Plasma exchange twice daily may be more effective if the desired outcome is not achieved. The authors specified that TTP (Moschcowitz’s syndrome) is a rare, urgent, life-threatening disease. The mortality rate in patients who are not treated appropriately and promptly is said to be as high as 80–90 %. Direct treatment reduces this to no more than 20 %, although orthopedic operations in particular may cause a much greater bleeding. The authors concluded that orthopedic surgeons and physicians of all specialties should be alert to the possibility of postoperative TTP [16]. Desmopressin may be used as a synthetic agent for treatment in mild forms. However, in severe type 1, types 2B and 2N, and type 3, or in patients who do not respond to desmopressin, the appropriate treatment is a factor VIII concentrate rich in VWF. Management in another study was based on erythrocyte and FFP transfusions with the administration of factor VII and accurate evaluation [8].
Conclusions
We recommend that careful preoperative checks for postsurgical bleeding risks be performed on the basis of three criteria: 1) family history (diathesis/hemophilia), 2) platelet counts (performed twice for definitive confirmation) and 3) coagulation tests such as prothrombin time, activated partial thromboplastin time, thrombin time, and international normalized ratio.
Received 04.08.2021
Revised 19.08.2021
Accepted 31.08.2021
Список литературы
1. Lavin M., O’Donnell J.S. How I treat low von Willebrand factor levels. Blood. 2019, Feb 21. 133(8). 795-804. doi: 10.1182/blood-2018-10-844936.
2. Mansour J., Graf K., Lafferty P. Bleeding disorders in orthopedic surgery. Orthopedics. 2012 Dec. 35(12). 1053-1062. doi: 10.3928/01477447-20121120-09.
3. Gopinath R., Sreekanth Y., Yadav M. Approach to bleeding patient. Indian J. Anaesth. 2014 Sep. 58(5). 596-602. doi: 10.4103/0019-5049.144664.
4. Diez-Zapirain M.A., Iruin Irulegui G., Rabanal A., Matorras R. Recurrent Hemoperitoneum after Oocyte Pick-Up in an IVF Patient with Von Willebrand Disease May be Prevented with a Combination of Factor VIII and Von Willebrand Factor. Int. J. Reprod. Med. Gynecol. 2019. 5(1). 1-3. doi: 10.37871/ijrmg.id38.
5. Heijdra J.M., Cnossen M.H., Leebeek F.W.G. Current and Emerging Options for the Management of Inherited von Willebrand Disease. Drugs. 2017 Sep. 77(14). 1531-1547. doi: 10.1007/s40265-017-0793-2.
6. Keesler D.A., Flood V.H. Current issues in diagnosis and treatment of von Willebrand disease. Res. Pract. Thromb. Haemost. 2017, Dec 12. 2(1). 34-41. doi: 10.1002/rth2.12064.
7. Higasa S., Tokugawa T., Sawada A. Diagnosis and management of von Willebrand disease. Rinsho Ketsueki. 2018. 59(10). 2222-2232. doi: 10.11406/rinketsu.59.2222.
8. Echahdi H., El Hasbaoui B., El Khorassani M., Agadr A., Khattab M. Von Willebrand’s disease: case report and review of literature. Pan. Afr. Med. J. 2017, Jun 29. 27. 147. doi: 10.11604/pamj.2017.27.147.12248.
9. Turkiz G. Von Willebrand Disease. Gazi University. Turk. Hematology Society. 2007. Basic Hemostasis Thrombosis Course Book. 2007. 52-59.
10. Macfarlane D.E., Stibbe J., Kirby E.P., Zucker M.B., Grant R.A., McPherson J. Letter: A method for assaying von Willebrand factor (ristocetin cofactor). Thromb. Diath Haemorrh. 1975, Sep 30. 34(1). 306-308.
11. Miller C.H., Graham J.B., Goldin L.R., Elston R.C. Genetics of classic von Willebrand’s disease. I. Phenotypic variation within families. Blood. 1979 Jul. 54(1). 117-136.
12. Rodeghiero F., Castaman G., Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987 Feb. 69(2). 454-459.
13. Bowie E.J., Fass D.N., Olson J.D., Owen C.A. Jr. The spectrum of von Willebrand’s disease revisited. Mayo Clin. Proc. 1976 Jan. 51(1). 35-41.
14. Nilsson I.M. In memory of Erik Jorpes. von Willebrand’s disease from 1926–1983. Scand. J. Haematol. Suppl. 1984. 40. 21-43.
15. Caviglia H., Landro M.E., Galatro G., Candela M., Neme D. Epidemiology of fractures in patients with haemophilia. Injury. 2015 Oct. 46(10). 1885-1890. doi: 10.1016/j.injury.2015.06.034.
16. Iosifidis M.I., Ntavlis M., Giannoulis I., Malioufas L., Ioannou A., Giantsis G. Acute thrombotic thrombocytopenic purpura following orthopedic surgery: a case report. Archives of Orthopaedic and Trauma Surgery. 2006 July. 126(5). 335-338. doi: 10.1007/s00402-005-0014-4.

/48.jpg)
/49_2.jpg)
/49.jpg)
/50.jpg)