
Газета «Новости медицины и фармации» №1 (782), 2023
Вернуться к номеру
Особливості стратегії поетапної нейропротекції закритої черепно-мозкової травми
Авторы: Черній В.І. (1), Нерянов К.Ю. (3), Середа Д.А. (2,3), Бєленічев І.Ф. (2,4), Кучеренко Л.І. (2,4)
(1) — ДНУ «Науково-практичний центр профілактичної та клінічної медицини» ДУС, м. Київ, Україна
(2) — Запорізький державний медико-фармацевтичний університет, м. Запоріжжя, Україна
(3) — КНП «9-та міська клінічна лікарня» ЗМР, м. Запоріжжя, Україна
(4) — ТОВ НВО «Фарматрон», м. Запоріжжя, Україна
Разделы: Справочник специалиста
Версия для печати
/6_u.jpg)
За даними ВООЗ, у світі щорічно трапляються черепно-мозкові травми (ЧМТ) більше ніж у 10 млн населення, з них 250–300 тис. постраждалих вмирають. У Європі ЧМТ є основною причиною смерті хворих віком до 35 років. Останнім часом частота ЧМТ в Україні в різних регіонах становила від 2,3 до 6 %, у середньому 4–4,2 %, тобто виникала у 200 тис. потерпілих протягом року.
Кожні 10–15 років частота ЧМТ зростає практично вдвічі [1–4]. ЧМТ має високу і неухильно зростаючу поширеність і найчастіше зустрічається в осіб молодого і середнього віку (20–50 років), тобто в найбільш працездатній групі населення. У великих промислових регіонах країни травматизм останніми роками набуває характеру епідемії.
Згідно з міжнародними епідеміологічними дослідженнями, щороку у світі від інсульту помирають 4,7 млн осіб. За даними літератури [5–8], частка летальних наслідків при крововиливах у мозок становить від 52 до 82 %, при субарахноїдальних крововиливах — від 32 до 64 %. Інсульт є основною причиною інвалідизації населення, тільки 20 % хворих, які вижили, повертаються до роботи [9–11].
Усе вищевикладене, а також різноманітність клінічних проявів і тяжкості перебігу ЧМТ обумовлюють необхідність розробки науково обґрунтованих підходів до патогенетичного лікування ЧМТ на основі поглибленого вивчення різних аспектів її патогенезу [9–14]. Актуальність проблеми лікування ЧМТ і її наслідків пов’язана зі значними економічними втратами, що доповнюються стійкою інвалідизацією 10–12 % постраждалих.
У патогенезі ЧМТ важливу роль відіграють вторинні пошкодження тканини головного мозку, центральне місце серед яких посідають нейрогіпоксія та церебральна ішемія, унаслідок чого формуються порушення енергетичного метаболізму, вуглеводного обміну, відбувається гіперактивація вільнорадикальних процесів, що ініціюють процеси переокиснення [1–4]. Вивчення особ–ливостей перебігу патофізіологічних процесів цереброваскулярних розладів (як гострих, так і хронічних) і травматичного пошкодження мозку свідчить, що в основі поширення порушень цілісності клітин і руйнування тканин при різних формах внутрішньочерепних гематом лежать механізми апоптозу або некрозу [5]. Сучасна медицина має великий асортимент лікарських засобів для лікування ЧМТ і запобігання її наслідкам, але далеко не всі препарати відповідають сучасним вимогам. Тому пошук і розробка нових високоефективних і безпечних засобів фармакологічного захисту мозку, що відзначаються політропністю фармакодинамічних ефектів, спрямованих на одночасну корекцію різних ланок патогенезу ЧМТ, є одним із пріоритетних напрямків розвитку фундаментальної та клінічної медицини.
Особливості патогенезу ішемічного ушкодження мозку
Останніми роками XX століття відбулася еволюція уявлень про механізм пошкодження тканини мозку при гостро виникаючій ішемії, що радикально змінило погляди на лікування гострого порушення мозкового кровообігу (ГПМК) [1–3, 17–30]. Тривалий час вважалося, що основним фактором, що ушкоджує, при церебральній ішемії будь-якого генезу є недостатнє постачання мозкової тканини енергетичними субстратами — киснем і глюкозою. Однак дані, отримані в 70-х роках минулого століття, показали, що ішемія (аноксія-гіпоксія) виступає тільки в ролі пускового фактора, що пошкоджує [27, 28]. Сучасна концепція патогенезу гострої ішемії мозку розглядає зміни його функції, пов’язані з редукцією мозкового кровотоку, реперфузією та формуванням зони ішемічної півтіні [28, 35].
У середині 80-х років було відкрито Са2+-індуковану ексайтотоксичність і розроблено теорію ексайтотоксичної смерті нейронів. Було встановлено, що зниження енергетичного потенціалу нервової тканини є тригерним механізмом, що зумовлює постаноксичні порушення метаболізму, іонного гомеостазу та активацію трьох основних механізмів аноксичного пошкодження клітин мозку: вільнорадикального, кальційзалежного і фосфоліпазного [28].
Аналіз динаміки патогенетичних механізмів, що запускаються гострою ішемією мозку, встановив їхню чітку часову послідовність. Протягом перших 3 годин із моменту ГПМК в ішемізованій тканині максимально вираженим є енергетичний дефіцит; через 3–6 годин розвиваються глутаматна ексайто–токсичність, порушення кальцієвого гомеостазу і лактат-ацидоз, що вщухають до кінця першої доби. Віддалені наслідки ішемії, такі як оксидантний стрес і локальне запалення, починають проявлятися через 2–3 години, досягають максимуму через 12–36 годин, а на 2-гу — 3-тю добу починається апоптоз. Ці процеси зберігаються тривало, протягом декількох місяців, сприяючи в постінсультному періоді прогресуванню процесів атерогенезу й дифузного пошкодження тканини головного –мозку, у тому числі енцефалопатії [32–35].
Встановлено, що ступінь ушкоджуючої дії ішемії визначається насамперед глибиною та тривалістю зниження мозкового кровотоку. Було сформульовано положення про критичний поріг мозкового кровотоку, коли припиняється метаболізм. Ділянка мозку з найбільш вираженою олігемією (менше за 10–15 мл/100 г/хв) протягом 6–8 хвилин стає необоротно пошкодженою. Це серцевина, або ядерна зона ішемії. Протягом кількох годин центральний точковий інфаркт є оточеним ішемізованою, але живою тканиною — зоною ішемічної півтіні, або пенумбри, у якій збережено енергетичний метаболізм на рівні підтримки функції клітини й розвиваються лише функціональні, але не структурні зміни. Ділянка ішемічної півтіні може бути трансформована в здорову тканину мозку і є головною мішенню терапії інсульту в перші години і дні захворювання [27, 28].
Патологічні зміни, що відбуваються при ішемії, називають ішемічним каскадом, сутність якого зводиться до наступного: зниження доставки О2 і глюкози викликає негайне розщеплення аденозинтрифосфату (АТФ) для покриття потреб клітин в енергії (триває 2–4 хвилини після повної ішемії). Далі використовується фосфокреатинін, рівень якого в мозку в 3 рази вищий за АТФ, для процесів ресинтезу АТФ з аденозиндифосфатів (АДФ). Розвивається зниження внутрішньоклітинного рН і перехід на анаеробний гліколіз, що веде до підвищення вмісту молочної кислоти, перетворення за допомогою лактату тривалентного заліза на двовалентне. Саме це сприяє утворенню вільних радикалів та окисненню ліпідів клітинних мембран [28, 38, 39].
Важливим моментом каскаду є підвищення рівня нейротрансмітерів — збуджуючих амінокислот глутамату й аспартату. Аспартат впливає тільки на N-метил-D-аспартат (NMDA) рецептори, які локалізовані в корі й гіпокампі. Глутамат активує три типи рецепторів: NMDA-рецептори, локалізовані в корі й таламусі, AMPA-рецептори, або рецептори до квізквалату, рецептори каїнату, що локалізовані в стріатумі гіпокампа. Активація NMDA-рецепторів сприяє входженню в клітину Na+, Cl–, Са2+ і H2О. Активація AMPA-рецепторів і рецепторів каїнату сприяє входженню в клітину Na+. Отже, зростання позаклітинної концентрації глутамату веде до загибелі клітин двома шляхами. Перший шлях — при активації глутаматом NMDA-рецепторів розвивається негайна нейротоксичність: при входженні в клітину Na+, Cl– і H2О виникає клітинний набряк, лізис мембран і клітинна смерть [27, 28]. Другий шлях — у період від 24 до 72 годин після ішемії формується відстрочена нейротоксичність — активація NMDA-рецепторів сприяє входу в клітину іонів Са2+, у результаті активуються фосфоліпази, протеази, вільні жирні кислоти, утворюються арахідонова кислота й вільні радикали, що веде до окиснення ліпідів і клітинної смерті.
Взагалі підвищене надходження іонів Са2+ у клітину — це рання й центральна подія ішемічного каскаду. Підвищений рівень внутрішньоклітинного кальцію –активує фосфоліпази, призводить до гідро–лізу мембранних фосфоліпідів і вивільнення жирних кислот, що спричиняє руйнування мітохондріальних і клітинних мембран. У підсумку набряк клітин, порушення проникності гематоенцефалічного бар’єра (ГЕБ) формують запалення [40, 41].
Сьогодні достатньо досліджена роль судинного ендотелію в патогенезі церебральної ішемії. Розвиток набряку мозку і постаноксичних ушкоджень ГЕБ значною мірою ініційований біологічно активними речовинами самої судинної стінки. Ішемізовані ендотеліальні клітини експресують і виділяють біологічно активні запальні цитокіни й хемокіни, адгезивні молекули й лейкоцитарні хемоатрактанти в зоні ГЕБ, стимулюють мобілізацію клітин крові та їх інфільтрацію в мозок. Пошкоджені мембрани клітин, точніше арахідонова кислота в місці деструкції нейроцитів, зсувають рН середовища в кислу сторону. Універсальні фагоцити — нейтрофіли мають властивість неспецифічного хемотаксису в бік кислої рН, тобто до місця ушкодження. Присутність у вогнищі пошкодження базофілів ускладнює ситуацію, збільшуючи проникність ГЕБ шляхом виділення гістаміну й гепарину при дегрануляції [28, 42, 43].
Ланцюг запальних реакцій триває протягом кількох днів навіть після відновлення перфузії тканин. Механізм лейкоцитарної міграції, дисфункції ГЕБ і посилення запального процесу посилюється тим, що нейрони, астроцити й ендо–теліальні клітини продукують протеїнази, які також неспецифічно пошкоджують гематоенцефалічний бар’єр. А нейтрофіли, що проникли в мозкову тканину, виробляють ще більше прозапальних цитокінів, у тому числі досить агресивний фермент циклооксигеназу-2 (ЦОГ-2), який вважається одним з ключових ферментів, що відповідають за запальну реакцію в центральній нервовій системі (ЦНС). Індукція ЦОГ-2 може відбуватися не тільки в нейронах, але й у макро–фагах, астроцитах, ендо–теліальних клітинах, що також призводить до пошкодження ГЕБ [28, 32].
Отже, набряк головного мозку виникає вже через кілька хвилин після розвитку локальної ішемії внаслідок пошкодження клітинних мембран і накопичення води в клітинах (цитотоксичний набряк), а також через пошкодження ГЕБ і потрапляння плазми в позаклітинний простір мозку (вазогенний набряк). Вираженість набряку мозку перебуває в прямій залежності від розмірів інфаркту мозку [5, 27].
Реалізація механізмів клітинної загибелі нейронів — некрозу й апоптозу
Встановлено, що в постгіпоксичному періоді реалізуються два механізми клітинної загибелі, які відзначаються специфічними морфологічними характеристиками, — некроз і апоптоз. –Однак сьогодні встановлено, що список механізмів клітинної смерті не є повним [1–5, 27, 28].
Послідовність патологічних подій при некрозі завжди подібна: осмоліз, спричинений клітинним набряком, призводить до пасивної смерті ушкодженої клітини. Одним із вторинних ефектів некрозу є запалення, яке викликається клітинним вмістом, що вивільняється, і супроводжується виробленням цитокінів. Як правило, некроз розвивається під дією екстремальної гіпоксії, коли не встигають вмикатися адаптаційні механізми [28].
Апоптоз, запрограмована клітинна загибель, є біологічно найбільш збереженою формою смерті клітини, вона використовується організмом для конт–ролю кількості, якості клітин і підтримки функціонування органів у процесі роз–витку [27, 35].
Співвідношення цих типів загибелі –нейронів за обсягом пошкоджених клітин залежить від ступеня й тривалості гіпоксії [28]. У низці випадків зазначено, що загибель нейронів іноді має риси і некрозу, і апоптозу. Основними елементами подібності між двома варіантами смерті нервових клітин є оксидативний стрес, підвищений вміст вільних іонів кальцію в цитоплазмі, активація протеаз. Явища розривів мембран нейронів, фрагментації їх ДНК, що характеризують відповідно некроз і апоптоз, описувалися іноді в тих самих уражених клітинах [27].
У той же час апоптоз і некроз мають чіткі відмінності. Насамперед апоптоз, енергозалежний процес, на відміну від некрозу, потребує АТФ. Також значущою відмінністю є часовий аспект: апоптоз, на відміну від некрозу, триває довше. І головне: клітинна загибель шляхом апоптозу дозволяє не пошкоджувати сусідні клітини й міжклітинний простір. Нейрон гине практично локально, з цілковитим збереженням навколоклітинного простору, і утилізує свої органели до молекулярних складових — амінокислот, СО2, Н2О. У зв’язку з цим спосіб активно спрямувати пошкоджені клітини на шлях апоптозу, а не некротичної загибелі є предметом пошуку багатьох дослідників, і механізми індукції нейронального апоптозу в умовах гострої церебральної ішемії інтенсивно вивчаються [1–5, 28].
У розвитку процесу апоптозу беруть участь безліч сигнальних молекул, багато з яких регулюють інші важливі функції організму. До найбільш вивчених факторів, здатних запускати в ней–роні програму апоптозу, належить оксид азоту (NO) — одна з ключових сигнальних молекул, що регулюють функції серцево-судинної, нервової та імунної систем організму [28]. Концентрація NO починає збільшуватися з перших хвилин ішемії, досягаючи максимуму на 1-шу — 3-тю добу. У початковому періоді ішемії превалює експресія конститутивної кальційзалежної NO-синтази (NOS), обумовленої транс–мітерним автокоїдозом. На цьому етапі NO бере участь у непрямих механізмах загибелі нейрона — активації фосфоліпаз, посиленні утворення гідроксил-радикала, модуляції активності NMDA-рецепторів. Починаючи з 7–14-ї доби при глобальній ішемії та з 1–3-ї доби при фокальній ішемії, тобто у відстроченому постішемічному періоді, реєструється гіперпродукція NO за участю індуцибельної NOS активованої глії, макрофагів і нейтро–філів [5, 21]. У численних роботах було показано безпосередню участь NO у процесі деструкції нейрона при ішемії при призначенні тваринам з гострими порушеннями мозкового кровообігу селективних інгібіторів нейрональної та індуцибельної ізоформ NOS, а також у дослідах на тваринах з дефіцитом гена, що кодує iNOS. Отримано дані про зростання концентрації NO у мозку тварин як з фокальною, так і з глобальною ішемією [5, 15, 17]. Концентрація NO починає зростати з перших хвилин ішемії, досягаючи максимуму на 1-шу — 3-тю добу. Вимірювання активності NOS показало різке підвищення активності цього ферменту як у вогнищі ішемії, так і в пенумбрі, однак без урахування належності до певного типу NOS. Участь NO в ушкодженні й загибелі нейрона має свою специфіку і визначається ізоформами NOS, а також видом і стадією розвитку інсульту. У початковому періоді ішемії превалює експресія конституційної кальційзалежної NOS, обумовленої трансмітерним автокоїдозом. Продукція NO на цьому етапі не є фактором, який безпосередньо визначає загибель нейрона.
Відстрочений характер експресії індуцибельної NOS пов’язаний з більш пізніми термінами появи активованої астро- і макроглії і клітин запалення. При фокальній ішемії вищезазначені клітини — продуценти NO знаходяться в пенумбрі, а при глобальній ішемії — в найбільш чутливих до дефіциту кисню структурах.
Крім NO-синтаз, джерелом NO в організмі теплокровних є нітрат-/нітритредуктази, здатні відновлювати нітрати й нітрити. Нітроредуктазну активність мають гліоцити й тимоцити. Відомо, що NO і його деривати можуть викликати перекисне окиснення фосфоліпідів. Під дією цитотоксичних дериватів NO і гідроксил-радикала відбувається відкриття мітохондріальних пор з експресією та виходом у цитозоль проапоптичних білків. Є дані про те, що NO прямо активує відкриття гігантської пори мітохондрії, що призводить до виходу цитохрому С і запуску каспазного каскаду. Відкриття пор перетворює мітохондрії з «електростанцій» на «топку» субстратів окиснення без утворення АТФ [28].
При цьому важливо підкреслити роль мікроглії, яка у великій кількості синтезує простаноїди, що утворюються з арахідонової кислоти циклооксигеназним шляхом, і NO, який синтезується з аргініну за допомогою індуцибельної NOS. Баланс між простаноїдами і рівнем NO може визначати напрямок мікрогліальної реакції нейропротекторним або нейро–токсичним шляхом [5, 28]. Співвідношення NO і тіол-дисульфідної системи є фактором, що визначає подальшу долю нейрона в умовах ішемії, а саме тип його загибелі. В умовах ішемічних ушкоджень головного мозку в ранні терміни розвивається нітрозуючий стрес, що призводить до нітрозування тіолів, змінюючи тіол-дисульфідну рівновагу білків мітохондріальної пори. На цій стадії мітохондріальна NOS відіграє захисну роль, регулюючи клітинну загибель, переключаючи її на більш вигідний тип — апоптоз. Далі розвиваються оксидативний і карбонільний стрес, які суттєво зміщують тіол-дисульфідну рівновагу в бік окиснених тіолів, розвивається стійка мітохондріальна дисфункція з дефіцитом енергетичних запасів клітини, розвитком автокоїдозу, зміною відповіді геному, і, як наслідок, клітина гине за типом некрозу [27]. Нейроапоптоз розвивається як каскадний процес, що супроводжується активацією специфічних про- або антиапоптичних білків, а також особливих протеолітичних ферментів — каспаз. Існують переконливі докази того, що центральна роль у продукції активних форм кисню і в подальшому розвитку апоптозу належить мітохондріям, зміні проникності їх мембран у результаті формування специфічного комплексу мітохондріальних пор та ініціації руйнування мітохондрій — мітоптозу [1, 28]. Мітоптоз — поліморфологічна форма загибелі мітохондрій, що може спричинити смерть усієї клітини шляхом апоптозу (внутрішній шлях за рахунок масованого вивільнення мітохондріальних субстратів). Цей механізм запускається при формуванні дисфункції мітохондрій. Існує узагальнене поняття «мітохондріальна дисфункція». Це типовий патологічний процес, який не має етіо–логічної та нозологічної специфічності. Розвиток мітохондріальної дисфункції призводить до порушення зворотного захоплення медіаторів (катехоламінів, дофаміну, серотоніну), іонного транспорту, генерації та проведення імпульсу, синтезу білка de novo, процесів трансляції та транскрипції; активізуються «паразитарні» енергопродукуючі реакції, що призводить до суттєвого зменшення енергетичних запасів нейрональної клітини. Відкриття пор відбувається за рахунок окиснення тіольних груп цистеїнзалежної ділянки білка внутрішньої мембрани мітохондрій (АТФ/АДФ-антипортер), що перетворює його на проникний неспецифічний канал-пору. У точних біо–хімічних дослідженнях було встановлено, що порушення кисневого режиму тканин, гіпер–продукція ексайтотоксичних аміно–кислот, зниження «нормальної» акумуляції Са2+ –мітохондріями, пошкодження мембрани мітохондрій АФК підсилює відкриття пор і вивільнення апоптогенних білків з пошкоджених мітохондрій. У цьому контексті суттєвою є роль одного з нейротрофічних факторів — фактора некрозу пухлини альфа (TNF-α), з яким пов’язане відкриття пор у мітохондріях, подальше порушення їх мембран і розвиток мітоптозу. Міто–хондріальна пора є каналом, що проходить через обидві мітохондріальні мембрани і складається з трьох білків: транслокатора аденінових нуклеотидів, потенціалзалежного аніонного каналу (порину) і бензодіазепінового рецептора. Це призводить до зниження мембранного потенціалу та набухання матриксу, цілісність зовнішньої мембрани неминуче порушується, і з міжмембранного простору до цитоплазми виходять білки апоптозу. Їх кілька: фактор, що індукує апоптоз (Apoptosis-inducing factor), вторинний міто–хондріальний активатор каспаз (Second mitochondria-derived activator of caspases — Smac) і деякі прокаспази. Фактор, що індукує апоптоз, прямує прямо в ядро, де викликає деградацію ДНК. Поряд зі специфічними апоптозними білками з мітохондрії через відкриту пору виходить цитохром С, який у нормі служить кінцевою ланкою електронтранспортного ланцюга. У цитоплазмі цей білок зв’язується з білком Apaf-1 (Apoptotic protease activating factor-1 — фактор 1, що активує протеазу) і формує апоптосомний комплекс. Він за допомогою Smac і ще одного фактора (Omi/HtrA2) активує прокаспазу-9, яка, ставши каспазою-9, перетворює два інші проферменти на каспази-3 і -7; а вони вже розщеплюють структурні білки, призводячи до появи біохімічних і морфологічних ознак апоптозу. Вважається, що мітоптоз — це механізм утилізації мітохондрій, у яких продукція активних форм кисню перевищила фізіологічний рівень. Вважається, що цей механізм загибелі клітин є оригінальним при серцево-судинній патології, особливо ішемічного характеру [1, 3, 27, 28].
У даний час серед механізмів програмованої загибелі нейронів, особливо при ЧМТ та ГІ, виділяють феро–птоз. Фероптоз — нещодавно описана форма запрограмованої загибелі клітин, що здійснюється за допомогою Fe-залежної генерації активних форм кисню під контролем глутатіонпероксидази і може бути інгібована хелаторами заліза і ліпофільними антиоксидантами. Цей вид загибелі був відкритий і описаний у 2016 році. Показано, що його індукторами можуть бути так звані малі молекули — ерастин, RSL3, FIN56. Один з розкритих молекулярних механізмів пов’язаний з тим, що при впливі ерастину інгібується робота цистеїн-глутаматного антипортера, який обмінює позаклітинний L-цистеїн на внутрішньоклітинний L-глутамат. Унаслідок цього кількість внутрішньоклітинного цистеїну різко знижується, це викликає дефіцит глутатіону, що синтезується з цистеїну. Виснаження запасу глутатіону призводить до загибелі клітини, що було показано в тому числі при експериментальному нокауті гена глутатіонпероксидази [28]. Морфологічно для фероптозу типовим є зменшення числа мітохондрій у клітині, ущільнення й руйнування їх крист і розрив зовнішньої мембрани. Очевидно, значення цієї форми також пов’язане з канцерогенезом, обумовленим порушенням роботи р53. Також показана індукція фероптозу при амінокислотному голодуванні з дефіцитом глутаміну і транспортного білка трансферину, зростанням концентрації заліза (II) і активацією реакцій Фентона і Габера — Вейсса.
Головна роль у загибелі нейронів зони ішемічної півтіні належить глутаматному каскаду й оксидативному стресу. При цьому одним з етапів є зміна характеру експресії генів — індукторів апоптозу і генів — інгібіторів апоптозу, порушення окисно-відновних процесів за рахунок блокади мітохондріального комплексу. Важливу роль у цьому відіграють так звані ранні гени сімейств c-fos [1, 3, 27, 28].
Так, в умовах гіперпродукції активних форм кисню нейрохімічними й біоенергетичними системами головного мозку в умовах ішемії головного мозку, а також при інших нейродеструктивних процесах відбувається активація експресії чутливих генів, багато з яких необхідні для захисту клітин від токсичних ефектів окиснювального стресу [3, 20]. Експериментальними роботами останнього десятиліття показано значну роль зміни характеру експресії генів раннього реагування c-fos у розвитку апоптозу нейрональних клітин при нейродеструктивних захворюваннях. Білок c-fos бере участь безпосередньо в процесі фрагментації ДНК та ініціюванні процесів апоптичної загибелі клітини [1].
Однією з первинних реакцій геному у відповідь на стрес різного генезу є індукція білків теплового шоку — Heat shock proteins (HSP), які називають відповідно до їх молекулярних мас. HSPs виступають як внутрішньоклітинні шаперони, які підтримують протеостаз клітин у нормі та в різних стресових умовах (гіпертермія, гіпоксія, оксидативний стрес, радіація тощо). Відповідно до молекулярної ваги в кілодальтонах (кДа) сімейство шаперонів розподіляється на кілька груп: низькомолекулярні/малі (у тому числі HSP27 і HSP40) HSP60, HSP70, HSP90 і HSP110. Хоча загальне завдання цих білків, що полягає в збережені життєдіяльності клітини так і залишається загальним, функції і тканино–спеспецифічність HSPs варіюють між групами у фізіологічних і стресових умовах [44].
Найбільший інтерес при ішемічному пошкодженні мозку викликає білок HSP70 як важливий компонент системи ендогенної нейропротекції, який у першу чергу виконує функцію внутрішньоклітинних шаперонів і забезпечує процеси фолдингу, холдингу й транспорту синтезованих білків, а також їх деградацію як в умовах нормоксії, так і при стрес-індукованій денатурації. Відомо, що до складу сімейства білків теплового шоку 70 входять індуцибельний/стресоподібний блок HSP72/HSP70i, конститутивний/фізіологічний білок HSP73/HSC70, конститутивний глюкозорегулюючий мітохондріальний білок GRP75, конститутивний ендоплазматичний білок GRP73, а також індуцибельна гемоксигеназа-1 (HO-1), яка бере участь у метаболізмі білірубіну [28, 44]. Конститутивна форма HSP70 присутня однаково в усіх субклітинних компартментах і бере участь у роботі систем життєзабезпечення клітин в умовах нормоксії. Навпаки, індуцибельна форма HSP70 з’являється в клітинах у відповідь на стрес, у тому числі при ішемічному інсульті [27, 28, 44].
У першу чергу HSP70 розглядається як білок-шаперон, що забезпечує потужний протеостаз клітин. Шаперонна функція HSP70 полягає у взаємодії з пошкодженими, денатурованими вперше синтезованими білками з наступним визначенням їх частки. Поява і накопичення в клітині пошкоджених білків збільшує експресію HSP70. Після термічного пошкодження, яке викликає денатурацію всіх білків, концентрація HSP70 може становити до 20 % від усіх цитоплазматичних білків. Дія HSP70 призводить до формування термотолерантності — феномену адаптації до дії підвищених температур і збільшення температурного порогу чутливості за рахунок стабілізації білкових молекул за допомогою HSP70 при початковому, менш сильному нагріванні [1, 44]. Білки HSP70 необхідні для відновлення й виживання клітин у стресових умовах, їх кількість корелює з тяжкістю перебігу різних патологічних станів, таких як ішемія, запалення, автоімунні процеси, злоякісні пухлини, бактеріальні та вірусні інфекції, нейродегенеративні захворювання. Участь HSP70 у механізмах холдингу, фолдингу й рефолдингу неправильно зібраних білків, а також дезагрегації і елімінації денатурованих білків, що накопичуються в умовах стресу, забезпечує захист мембран органел, передусім мітохондрій, і самих клітин від пошкоджень [1, 3, 28, 44]. У відповіді на ішемію реєструється різке зростання рівня HSP70, при цьому його найбільша концентрація спостерігається в життєво важливих частинах клітин: ядерному, навколоядерному просторі, мітохондріях, ендоплазматичному ретикулумі, що свідчить про важливість шаперону-70 у захисті від загибелі. У міру відновлення роботи ядерних прерибосом концентрація HSP70 зменшується в ядрі й підвищується в цитоплазмі клітин. Отже, рівень HSP70 може розглядатися як маркер клітинного й тканинного пошкодження. Гіперпродукція в клітинах HSP70 інгібує розвиток автофагії як альтернативний, більш «радикальний» механізм клітинної відповіді на стрес [44].
Науковими роботами останніх років встановлена пряма цитопротекторна дія HSP70, яка реалізується шляхом регулювання процесів апоптозу й некрозу клітин. HSP70 гальмує мітохондріальний і цитоплазматичний шлях апоптозу. Так, HSP70 пригнічує перехід прокаспази-9 в активну каспазу-9 і порушує формування апоптосоми в цитоплазмі клітин. На тлі гіпер–експресії HSP70 підвищується рівень антиапоптичного білка Bcl-2 (B-cell lymphoma 2), який перешкоджає виходу цитохрому з мітохондрій і транслокації фактора, що індукує апоптоз.
Активація мікрогліальних продуктів у ділянці пенумбри призводить до транзиторної активації генів, що кодують фактори транскрипції (у тому числі c-fos) у перші кілька хвилин від початку інсульту. Далі запускається друга хвиля експресії генів білків теплового шоку (HSP70 і HSP72), що наростає протягом перших 1–2 годин захворювання і знижується в 1-й — 2-й дні [1, 27].
Особливості репаративних процесів нервової тканини
Дослідження останніх років істотно змінили уявлення про нейрон як нездатний до регенерації, що швидко й необоротно гине при впливі ішемії та гіпоксії. З’ясувалося, що загибель диференційованих нейронів при пошкодженні їх аксонів не така вже й неминуча [5, 27, 30].
По-перше, встановлено, що мозок містить стовбурові клітини, які самовідновлюються і з яких можуть формуватися попередники нейронів, астроцитів і олігодендроцитів, що здатні мігрувати, диференціюватися й інтегруватися в мозку [6]. По-друге, нейрони виявилися значно стійкішими до таких впливів, як ішемія та гіпоксія. Особ–ливо якщо на тлі впливу здійснюється нейропротекція (гіпотермія, блокада ексайтотоксичних рецепторів). У цих умовах ушкодженню й загибелі нейронів протистоять процеси проліферації та репарації [7, 27].
Отже, у ранньому постгіпоксичному періоді некрозу піддаються лише найбільш чутливі й не захищені нейро–трофічними факторами нейрони, інші піддаються апоптозу або функціонують завдяки активації систем репарації ДНК. Процеси репарації можуть бути посилені застосуванням нейротрофічних факторів, трансплантацією нейронів, використанням методів генної інженерії [8, 28, 44]. Досконалість організації ЦНС підтверджується наявністю захисних механізмів підтримки структурно-функціо–нального стану нейронів, до яких належать –нейротрофічність, нейропротекція і –нейропластичність. Нейротрофічність — природний процес, що полягає в проліферації, диференціації та виживанні нервових клітин. Нейропротекція, безперервна адаптація нейрона до нових функціональних умов, є сумою всіх механізмів, спрямованих проти ушкоджуючих факторів. Нейропластичність включає відновлення функцій після природних пошкоджень та інших порушень, викликаних будь-якими агентами. Ці три фундаментальні біологічні процеси не мають чітко виражених меж. Вони переплетені між собою і разом забезпечують збереження та регенерацію нервової тканини [28]. Однак ці захисні механізми можуть бути активовані як природними факторами, так і фармакологічно [5, 7, 8, 11]. Тому фармакологічна корекція, спрямована на обмеження нітрозуючого, оксидативного й карбонільного стресів, дозволяє знизити кількість деструктивно змінених клітин, а також переключити тип загибелі клітини з некрозу на апоптоз [28].
Так, наприклад, на відміну від ушкоджуючого ефекту надмірної та тривалої активації NMDA-рецепторів, їх фізіо–логічна стимуляція в синапсах сприяє виживанню нейронів, активуючи нейро–трофічні й нейропластичні процеси [1, 27]. Глутамат відіграє важливу роль у процесах диференціювання і життєздатності нейронів, переважно через посилення вхідного струму Са2+, у той час як значне зниження активності NMDA-рецепторів in vivo призводить до поширеного апоптозу серед клітин, що розвиваються, посилює поточні –нейродегенеративні процеси, тобто знижує можливість виживання клітин в умовах ішемії [28].
Встановлено, що нейрони, крім трансмітерів, можуть синтезувати нейро–трофічні фактори й цитокіни, які антеградно й ретроградно транспортуються аксонами і впливають на синтез, вивільнення й ефективність дії трансмітерів. Нейро–трофічні речовини змінюють функціональну активність нейронів, які мають рецептори для цих класів речовин [5, 28]. Відзначається функціональне злиття –нейротрансмітерної, нейротрофічної та нейроімунної регуляції в єдину систему, де нейротрансмітери можуть діяти як трофічні речовини й імунорегулятори, і навпаки, нейротрофіни й цито–кіни можуть або безпосередньо впливати на активність нейронів, або модулювати дію нейротрансмітерів. У результаті той самий сигнал може радикально змінювати характер дії залежно від низки додаткових впливів — пресинаптичних впливів, об’ємної передачі, паралельної дії –нейротрофінів, цитокінів і нейрогормонів. Також встановлено, що астроцити через відповідні рецептори постійно отримують інформацію про стан нейронів, беруть участь у синтезі та інактивації нейротрансмітерів, синтезують нейротрофіни й цитокіни, захищають нервові клітини від загибелі [1, 3, 27].
Отже, сьогодні не викликає сумнівів необхідність розробки та початку з перших днів захворювання репаративної терапії, спрямованої на поліпшення пластичності здорової тканини, що оточує ділянку інфаркту мозку, активацію утворення полісинаптичних зв’язків, збільшення щільності рецепторів [27, 28].
Патогенетична єдність механізмів ішемічного і травматичного ушкодження мозку
Сучасні дані про патофізіологію мозкового інсульту й пошкодження мозку внаслідок черепно-мозкової травми свідчать про патогенетичну єдність механізмів клітинного пошкодження при будь-якій гострій церебральній недостатності, що обумовлено тканинною ішемією, яка обов’язково виникає [4–13].
Будь-які ушкодження головного мозку (вогнищеві або дифузні, транзиторні або постійні) клінічно проявляються цереброваскулярними розладами. І хоча інсульт і травматичне пошкодження мозку виникають унаслідок різних причин, в основі загибелі нервових клітин лежать однакові молекулярні й клітинні механізми [5, 7, 8, 11, 28]. Відповідно до сучасних уявлень про патогенез ушкоджень і захворювань головного мозку виділяють дві групи чинників, які впливають на перебіг і прогноз при такій патології: первинне ураження і так звані вторинні ушкодження ішемічного характеру [28]. При ішемічному й травматичному пошкодженні головного мозку навколо вогнища ушкодження формується зона ішемічної пів–тіні — пенумбри. Ішемія у своєму розвитку має 2 стадії — біоенергетичної гіпо–ксії, або стадії оборотних змін, і метаболічної гіпоксії, або необоротних змін [28].
Однотипно розвивається реакція гліальних клітин на ушкоджуючий фактор (травму, ішемію, крововилив) з розвитком дисбалансу цитокінів, локальною запальною реакцією, що веде до пошкодження нейронів, ГЕБ і порушень мікроциркуляції [5].
Гіперактивність системи NMDA-рецепторів, оксидативний стрес, як і продукція прозапальних цитокінів гліальними клітинами, є основними патогенетичними чинниками формування набряку мозку й індукування процесів апоптозу нейронів [44].
Нейрозапалення — це процес, що розвивається як при травмах, так і при інсультах. При ЧМТ запалення є закономірним механізмом, особливо при відкритій ЧМТ [5, 28]. Розрізняють гостру й хронічну стадії запалення, з початковою інфільтрацією нейтрофілами та макрофагами, а пізніше — із залученням і інших типів клітин [27]. Роль цитокінів при ЧМТ складна, адже вони мають як негативний, так і позитивний ефект. Так, наприклад, TNF-α, що ускладнює швидку відповідь організму на запалення, пізніше бере участь у регенеративній фазі відновлення. Інтерлейкін-6 на ранніх стадіях ушкодження бере участь у захисті від запалення, а пізніше його гіперсинтез астроцитами призводить до загибелі нейронів і розвитку неврологічної симптоматики [5, 7]. Отже, будь-яка стратегія боротьби із запаленням у мозковій тканині має враховувати можливу двоїстість механізмів відповіді на ушкодження.
Церебральна ішемія також швидко призводить до розвитку запального процесу в головному мозку, а саме до інфільтрації мозкової тканини лімфоцитами і продукції цитокінів. Загибель нейронів відбувається переважно в зоні ураження. Як і при ЧМТ, спостерігається підвищений рівень прозапальних цитокінів після ішемічного ушкодження [5].
Встановлено, що в результаті пошкодження головного мозку внаслідок ЧМТ, геморагічного інсульту (ГІ), ішемічного інсульту (ІІ) у всіх хворих, незалежно від виду й результату мозкового пошкодження, у найгострішому періоді (1-ша — 10-та доба) формуються неспецифічні пост–агресивні синдроми зі стереотипним перебігом [1, 13, 14, 28]. Найбільш стійкими, клінічно й прогностично значущими в цьому періоді є синдром пристосувальної артеріальної гіпертензії, пристосувальної гіпернатріємії, запальний синдром, синдром гемостазіологічних порушень, гіперферментемія та гіперлактатемія, катаболічний синдром. Кількісні та якісні характеристики синдромів однотипні у хворих з ЧМТ, ГІ та ІІ і різняться тільки за наслідком і ступенем ушкодження мозку. Неспецифічні постагресивні синдроми у хворих з несприятливими наслідками ЧМТ, ГІ та ІІ мають ідентичні морфологічні еквіваленти, що виявляються при патолого-анатомічних дослідженнях, які можна порівняти за ступенем вираженості, поширеності, локалізації та частоти виникнення.
Постагресивні реакції зберігають адаптаційний потенціал до 5 діб (точка неповернення). Після цього терміну надмірна напруга компенсаторних механізмів призводить до їх виснаження. Оптимальна тривалість максимальної напруги компенсаторних реакцій у хворих на ЧМТ, ГІ та ІІ становить 3 доби. Саме в цей термін інтенсивна терапія має найбільшу ефективність.
Спільність патогенезу й танатогенезу, що обумовлена стереотипністю перебігу неспецифічних синдромів, дозволяє розглядати хворих на ЧМТ, ГІ та ІІ як єдину групу нейрореанімаційних хворих, що робить можливим і необхідним уніфікацію та стандартизацію лікувально-діагностичних і прогностичних підходів до ведення хворих з мозковими ураженнями в найгострішому періоді [1, 27, 28, 35].
Нами встановлено, що моделювання ЧМТ та ГІ у щурів супроводжується зміною характеру експресії білків casp3, NeuN (нейрональний ядерний антиген знижується при гіпоксії та травмі головного мозку) і GFAP (фібрилярний кислий білок, що є маркером пошкоджень гліальних клітин). Характер експресії залежить від тривалості ЧМТ і ГІ, а також від експериментальної терапії, що проводиться.
Експресія білків casp3 відображає посилення процесів клітинної загибелі в гіпокампі в ранньому періоді ЧМТ і ГІ, що документується його динамікою в контрольних групах дослідження — значне зростання експресії casp3 на ранніх термінах ЧМТ і ГІ (4-та доба) з поступовим зниженням на 7-му добу. Посилення процесів клітинної загибелі також підтверджується рівнем експресії NeuN (зниження на 4-ту і 7-му добу експериментальної ЧМТ і ГІ) у всіх групах спостереження. Посилення експресії GFAP (у 12 і 9 разів на 4-ту і 7-му добу) при ЧМТ і ГІ відображає процес активації астроцитів, що може мати як негативний, так і позитивний вплив на процеси відновлення, враховуючи наявність двох різних типів реактивних астроцитів.
Дослідження функціонального стану ураженого головного мозку
Для вивчення динамічних аспектів взаємозв’язку процесів, які відбуваються в мозку, використовують методи функціо–нальної візуалізації, засновані на новітніх технологіях. Такі методи, як функціо–нальне магнітно-резонансне сканування, позитронно-емісійна томографія і комп’ютерна однофотонна томографія, дозволяють спостерігати різноманітні форми активності в різних галузях мозку людини під час вирішення різноманітних когнітивних завдань [5]. Незважаючи на інтенсивний розвиток методів «функціо–нальної» нейровізуалізації з детальним дослідженням кровообігу мозку і його метаболізму, адекватну інтегральну оцінку стану ЦНС можна отримати лише за допомогою електроенцефалографічного дослідження [5–14].
Сучасна електроенцефалографія належить до найбільш поширених методів вивчення діяльності головного мозку людини в нормі та патології. Неінвазивність і безпека, відсутність обмежень за часом дослідження та станом людини, менша вартість порівняно з рентгенологічними методами нейровізуалізації є безперечними перевагами даного методу.
При візуальному аналізі електроенцефалограми (ЕЕГ) можна характеризувати морфологію хвиль, їх частоту, амплітуду, характер розподілу по корі, і таким чином можна скласти уявлення про дифузні й локальні зміни на ЕЕГ. Найчастіше цього буває достатньо для оцінки функціонального стану мозку. Однак при тривалому динамічному спостереженні цей процес стає дуже трудомістким.
Впровадження методів математичного аналізу суттєво підвищило інформативність ЕЕГ. Значну допомогу надає використання комп’ютерних програм. Спектральний аналіз відображає повний спектр складових ЕЕГ-ритмів, у тому числі тих, які не видно при візуальній оцінці, хоча великі похибки до методу вносять артефакти запису, які зазвичай постійно присутні в ЕЕГ, особливо при тривалій її реєстрації в умовах операційної або палати інтенсивної терапії [5–8].
Цих дефектів не позбавлений і найпоширеніший метод компресійного спектрального аналізу за Фур’є, заснований на перетворенні складових ЕЕГ-коливань і графічному їх розкладанні на ряд гармонічних частот. Перевагою даного методу є його доступність, можливість тривалої реєстрації спектрів, подання в стислій формі, можливість кількісної оцінки результатів і їх зіставлення [28].
Поряд зі спектральними методами аналізу ЕЕГ, що дають її амлітудно-частотну характеристику, при оцінці функціонального стану мозку використовують методи кореляційного аналізу. Вони відображають співвідношення ритмів ЕЕГ між окремими точками мозку всередині однієї півкулі, або між півкулями (кроскореляція, когерентність), або між різними епохами та за різними ритмами в одному відведенні (автокореляція, бі–спектральний індекс). Ці методи характеризують порушення організації електричних процесів мозку. Спектрально-когерентний аналіз набув значного поширення, а його функціональна значимість була доведена в роботах багатьох авторів [5–12, 27, 28].
Останнім часом у клінічній нейро–фізіології набув поширення метод просторового картування біоелектричної активності мозку. Він дозволяє подати ЕЕГ у вигляді кольорових карт, дати характеристику складових її ритмів і їх взаємодії, виклавши її у цифрових параметрах. У даний час цей метод аналізу використовується переважно для вирішення наукових завдань у нейрофізіології, неврології та інтенсивній терапії неврологічних хворих.
Діагностична цінність ЕЕГ ґрунтується на описі вираженості фізіологічних ритмів і їх співвідношень з подальшою інтерпретацією, що спирається на величезний теоретичний і практичний матеріал. Ми вважаємо, що значення діагностичних комплексів, заснованих на аналізі ЕЕГ, важко переоцінити. Інформативність ЕЕГ у клініці сьогодні базується на зіставленні патерну з особливостями клінічної картини, перебігу захворювання, на складних статистичних зіставленнях кількісних характеристик ЕЕГ і даних комплексного обстеження, включно із сучасними методами нейровізуалізації — КТ і МРТ [5–11]. Такий підхід дозволяє максимально об’єктивно оцінювати функціональний стан ЦНС з точки зору цілісності та здатності до діяльності –нейронів, а також когнітивних функцій мозку в цілому.
Основні напрямки нейропротекції при гострій церебральній недостатності
Пріоритетним завданням для дослідників у цьому напрямі є пошук методів, які збільшують толерантність мозкової речовини до гіпоксичного ушкодження. Інтенсивна терапія гострих цереброваскулярних розладів включає три основні групи заходів [27, 28]: 1) неспецифічні, або загальні заходи для підтримки вітальних функцій; 2) специфічні заходи, спрямовані на поліпшення перфузії (внутрішньовенний або внутрішньоартеріальний тромболізис); 3) нейропротекція — превентивні заходи (антикоагулянти, антиагреганти, статини, хірургія сонних артерій).
Нейропротекцію поділяють також на пряму (застосування лікарських засобів або фізіологічних впливів, що інгібують біохімічні, метаболічні та клітинні наслідки ішемічного пошкодження) і непряму (забезпечення відновлення кровотоку й доставки енергетичних субстратів до ішемізованої, але не загиблої тканини мозку) [13].
Більш широке визначення нейропротекції включає застосування лікувальних підходів, що знижують частоту виникнення нових і/або повторних інсультів, зменшуючи обсяг і вираженість ушкодження мозку в разі розвитку інсульту, це так звана профілактична нейропротекція [1, 28].
Цитопротекція — це своєчасні й спрямовані впливи на системний і локальний рівні ушкодження (церебральний, кардіальний, печінковий тощо), викликані чинниками патогенезу захворювання, з метою запобігання необоротним змінам функціональних систем, які підтримують гомеостаз організму [5].
Раціональна нейроцитопротекція при гострій і хронічній ішемії мозку — це вплив на різні етапи ішемічного каскаду для створення умов, що адаптують нейрони до нових функціональних умов [27]. Основні механізми впливу нейроцитопротекторів при гострому інсульті включають блокаду повільних Са2+-каналів, конкурентний антагонізм до NMDA-рецепторів, збереження композиції нейрональних мембран за рахунок стабілізації їх фосфоліпідних структур, використання енергетичних ресурсів проміжного обміну речовин, що лежить в основі антигіпоксичної дії антиоксидантів [44].
Дослідники вважають, що для своєчасної корекції гіпоксичного стресу у вигляді метаболічного, оксидативного і медіаторного дисбалансів необхідно певним чином комбінувати цитопротектори різних класів [28, 44].
Однак невдачі, що виникли при клінічних дослідженнях нових препаратів, призвели до того, що в посібниках з лікування інсульту або не згадується зовсім про нейропротекцію, або дуже коротко констатується, що для її застосування на практиці немає підстав, оскільки клінічні дослідження не підтвердили її ефективності. Більшість лікарських засобів, що успішно застосовувалися в експериментальних дослідженнях, виявилися неефективними в клінічних дослідженнях, за винятком внутрішньовенного тромболізису, який показаний у 5–15 % ішемічного інсульту, і деяких нейропротекторів (цитиколін).
Сьогодні вивчення питання про нейро–протекцію дозволяє виділити два методологічно різних її види: нефармакологічну і фармакологічну. До нефармакологічної відносять гіпотермію, запобігання гіперглікемії, нестабільності гемодинаміки, гіпоксемії та гіперкапнії, гемодилюцію, нормалізацію підвищеного внутрішньочерепного тиску, корекцію кислотно-основного й електролітного дисбалансу. Для фармакологічної нейропротекції використовують препарати, які як зменшують активність обміну речовин у клітині, так і впливають на ланки ішемічного каскаду [1, 5, 27, 28, 44].
Разом з тим деякі роботи показали, що клінічна ефективність нейропротекторних препаратів, дія яких спрямована на сіру речовину, при підкіркових інсультах знижується. Імовірно, у цей момент в ішемізованій тканині мозку знижено кількість рецепторів — точок застосування протекторів. Тобто нейрони, для підтримки яких доставлено фармакологічний препарат, були не в змозі його акцептувати. Можливо, це і є основною причиною неуспіху існуючих нейропротекторних стратегій [5].
Також відсутність позитивної доказової бази при проведенні клінічних досліджень могла бути пов’язана з виявленими помилками чи недоліками у виборі доклінічних і клінічних досліджень. Більше того, є думка, що проводити рандомізовані плацебо-контрольовані подвійні (потрійні) сліпі дослідження в галузі інтенсивної терапії не має сенсу через неможливість їх уніфікації [37, 44].
І лише успіхи фундаментальних досліджень ушкоджень мозку зародили надію на те, що лікувальні впливи, спрямовані на усунення або пригнічення тих чи інших ланок патогенезу, можуть запобігти пошкодженню тканини мозку і, отже, знизити летальність внаслідок інсульту або травми, а також зменшити зумовлену ними інвалідність [1–30].
Практичні рекомендації щодо лікування ЧМТ засновані на трьох рівнях доказів: 1-й — стандарт, який необхідно застосовувати обов’язково; 2-й — рекомендації, які використовувати бажано; 3-й — опційна інформація, яка застосовується на розсуд [5, 28].
Аналізуючи велику кількість сучасних протоколів з лікування ЧМТ з позицій доказової медицини в сучасній нейро–травматології, необхідно зазначити, що через недостатню кількість досліджень 1-го і навіть 2-го класу рекомендації на рівні стандартів сформульовані тільки для розділів гіпервентиляції, застосування глюкокортикоїдів і ролі протисудомної терапії. Для інших пунктів — сформульовані на рівні опцій (варіантів, думок). Що стосується застосування –нейропротекторів, то рекомендації щодо їх застосування в сучасних посібниках з інтенсивної терапії тяжкої ЧМТ відсутні [28]. У літературі широко обговорюються можливості фармакологічної нейропротекції при ЧМТ. Публікації, як правило, відображають досвід застосування нейропротекторів для компенсації симптому або симптомокомплексу. Причому думки щодо використання нейропротекції при ЧМТ надзвичайно суперечливі — від ефективності в одних пацієнтів до відсутності позитивної динаміки або навіть погіршення в інших [6, 7, 27].
На підставі доказів 1-го рівня встановлено, що застосування гормонів при ЧМТ збільшує летальність на 4 %, тому вони не рекомендуються до застосування. З доказів 2-го рівня прийняті рекомендації щодо таких положень: необхідно уникати гіпотензії та підтримувати систолічний артеріальний тиск понад 90 мм рт.ст., уникати профілактичної гіпервентиляції, підтримувати внутрішньочерепний тиск (ВЧТ) лише на рівні трохи понад 20 мм рт.ст.
Зазначено, що показник ВЧТ набув останніми роками вагомого значення і став, по суті, наріжним каменем у терапії тяжкої ЧМТ. Доведена важливість постійного моніторування ВЧТ і церебрального перфузійного тиску [9, 28].
Останніми роками утвердився підхід до гіпотермії як до методу нейропротекції, що зменшує рівень мозкового метаболізму, ступінь неврологічних ускладнень ЧМТ, руйнування тканини, аксональне пошкодження, набряк, мікросудинну дисфункцію [17]. При зниженні температури у хворих на ЧМТ до рівня 32–35 °С понад 48 годин знижується смертність (3-й рівень доказів).
3-й рівень доказів стосується профілактики інфекційних ускладнень при тяжкій ЧМТ, профілактики венозних тромбозів, які при тяжкій ЧМТ зустрічаються у 20 % спостережень, гіпер–осмолярної терапії.
Останні дані метааналізу показали відсутність несприятливих реакцій на введення альбуміну, більше того, встановлено, що 20% гіперонкотичний розчин альбуміну покращує органну функцію, що оцінюється за шкалою SOFA [28].
Слід зазначити, що постійно ведуться розробки нових напрямів інтенсивної терапії ЧМТ. Наприклад, декомпресійна краніотомія — це метод, з приводу якого нині немає однозначної думки. За останніми даними, розмір краніотомії повинен бути не менше ніж 15 x 15 см [28].
У великому дослідженні, проведеному Американським національним інститутом неврологічних хвороб та інсульту, було зроблено спробу комбінування 6 найбільш успішних нових нейротропних препаратів з комплексом класичної терапії. Наголошується на важливості проведення повного комплексу лікування в перші 72 години після травми.
З особливостей ефективного застосування препаратів привертає увагу успішне використання еритропоетину при тяжкій ЧМТ, показане австралійськими й американськими дослідниками. На їхню думку, еритропоетин проникає через гематоенцефалічний бар’єр і чинить нейропротективну дію на ЦНС, знижує смертність у пацієнтів із травмою, його терапевтичне вікно становить 6 годин [28].
У даний час з’явилися докази нейропротекторних властивостей статинів, які не пов’язані з дією на холестерин. Новий препарат у списку нейропротекторів — це прогестерон, який захищає і відновлює гематоенцефалічний бар’єр, зменшує церебральний набряк. Показано нейро–протективну активність тамоксифену в дозах, що зумовлюють його міметичну дію щодо β-естрогенових рецепторів нервової тканини. Подібна дія модуляторів естрогенових рецепторів пояснюється, по-перше, їх прямими анти–оксидантними ефектами, а по-друге, підвищенням внутрішньоклітинної концентрації HSP70 за рахунок активації β-естрогенових рецепторів і від’єднання від останніх білків теплового шоку (hsp), що забезпечує проникнення hsp всередину клітини HSP70 [2]. Є дані про захисну роль HSP70 при церебральній ішемії, що супроводжується інтенсифікацією процесів вільнорадикального окиснення, зміщенням тіол-дисульфідної рівноваги, розвитком нітрозуючого стресу, глутаматної ексайтотоксичності [44].
Експериментально доведено нейропротективну дію Ангіоліну ((S)-2,6-діа–міногексанової кислоти 3-метил-1,2,4-триазоліл-5-тіоацетат), розробленого в НВО «Фарматрон». Нейропротективна дія Ангіоліну спрямована на гальмування нейроапоптозу, оксидативного й нітрозативного стресу, нормалізацію окисного метаболізму в головному мозку при ЧМТ, ІІ та ГІ. Призначення Ангіоліну приводить до підвищення виживання ендо–теліоцитів судин капілярної мережі кори головного мозку й судинної стінки судин мозку, підвищує кількість ендотеліоцитів, що проліферують, і підвищує експресію васкулоендотеліального фактора (VEGF). Ангіолін підвищує експресію HSP70 у цито–золі й мітохондріях нейроцитів і приводить до збільшення кількості мітохондрій з ознаками порушень ультраструктури. Ангіолін (розчин для ін’єкцій) успішно пройшов першу стадію клінічних досліджень.
Загалом для сучасної нейротравматології загальними принципами лікування є: адекватна оксигенація, перфузія, харчування, глікемічний контроль і нормотермія. Не рекомендується гіпервентиляція для контролю ВЧТ, яка підсилює ступінь ішемії. Осмотерапія, як і раніше, є загальноприйнятим способом регуляції ВЧТ, що довів свою ефективність, але не замінює декомпресійну краніотомію, яка повинна проводитися якомога раніше.
Загальна думка дослідників полягає в тому, що результат лікування пацієнтів із ЧМТ залежить від віку, неврологічного статусу, тяжкості травми, якості догляду, організації догоспітальної допомоги, рівня інтенсивної терапії [27, 28, 44]. Отже, ще раз наголошується, що серед величезної кількості фармакологічних засобів і загальних принципів терапії ЧМТ підбір препаратів для нейропротекції має ґрунтуватися на індивідуальній схемі, що базується на об’єктивній оцінці статусу та стану кожного пацієнта.
Визначальну роль в ангіоневро–логії стала відігравати сучасна концепція гетеро–генності ІІ, сформульована на початку 90-х у нашій країні. В основі цієї концепції лежать уявлення про різноманітність причин і механізмів розвитку гострого вогнищевого ішемічного ушкодження мозку. Прийнято виділяти атеротромботичний, гемодинамічний, кардіоемболічний, лакунарний інфаркт, інфаркт на кшталт гемореологічної мікро–оклюзії, а також гостру гіпертонічну енцефалопатію [28]. Сучасна тактика лікування ішемічного інсульту враховує основні причини його виникнення, механізм розвитку й особливість перебігу патологічного процесу, хоча надзвичайна короткочасність терапевтичного вікна для ядра інфаркту та періінфарктної ділянки суттєво знижує ефективність лікування пацієнтів [5, 7, 27].
Існуюча концепція лікування ішемічного інсульту передбачає два основних напрями: максимально раннє відновлення мозкового кровотоку й нейропротективну терапію [28].
Реперфузія ставить за мету відновлення або посилення кровотоку судинами в ділянці пошкодження і здійснюється шляхом тромболізису, вазодилатації, збільшення перфузійного тиску й поліпшення реологічних властивостей крові [27, 28]. Препаратом вибору є тканинний активатор плазміногену (tPA). На жаль, обмеження застосування тромболітичної терапії насамперед пов’язане, як уже згадувалося, з короткою тривалістю терапевтичного вікна. Уже через 3 години від початку ішемічного інсульту переваги від застосування tPA суттєво зменшуються, перекриваючись стрімко зростаючим ризиком геморагічних ускладнень. Тому перспективним є такий новий тромболітик, як десмотеплаза, який може застосовуватися в межах перших 9 годин після початку інсульту. Однак у рутинній практиці клінічні переваги від реперфузії можуть отримати менше ніж 5 % пацієнтів, які постраждали від інсульту [28].
Що стосується нейропротективної терапії, то класична ідеологія лікування ішемії мозку, уявлення про пенумбру, глутаматну ексайтотоксичність тощо передбачає застосування первинної та вторинної нейропротекції.
Причому первинна нейропротекція спрямована на переривання швидких реакцій глутамат-кальцієвого каскаду, вільнорадикальних механізмів; вона починається з перших хвилин ішемії та триває протягом трьох днів. А вторинна нейропротекція спрямована на блокаду прозапальних цитокінів, молекул клітинної адгезії, гальмування прооксидантних ферментів, відновлення нейротрофіки і переривання апоптозу. Вторинна нейро–протекція може бути розпочата через 6–12 годин після початку ішемії та триває не менше за 7 діб [27, 28].
Здавалося б, з точки зору патогенезу ішемії та фармакокінетики препаратів проблема купірування і лікування ГПМК має бути вирішена, проте клінічні випробування, що перевіряють безліч потенційних нейропротекторних речовин, дали негативні результати або такі, що розчаровують [28]. Метааналіз міжнародних досліджень нейропротекції, проведений у США, виявив лише два нейропротектори — церебролізин і цитиколін, які відповідають критеріям доказової медицини [28].
Європейське мультицентрове дослідження PASS I (Piracetam Acute Stroke Study) свідчить про ефективність пірацетаму при інсульті [1, 5, 28].
Деякі дослідники вважають, що найбільший інтерес для вивчення в цьому напрямі становлять такі групи препаратів, як антагоністи глутамату, антиоксиданти, блокатори кальцієвих каналів, хелатори кальцію та деякі інші.
Отже, на сьогодні визначено й обґрунтовано деякі принципи надання невідкладної допомоги хворим з порушеннями мозкового кровообігу. Лікування пацієнтів з ІІ має включати такі позиції: загальний моніторинг (контроль артеріального тиску й температури, рівня глікемії та іонного складу крові), специфічне лікування в гострій фазі (інтравенозний або інтраартеріальний тромболізис), превентивну терапію (антикоагулянти, антитромбоцитарні препарати, хірургічне втручання на каротидних судинах) [28].
Щодо ГІ, то медикаментозних методів його лікування нині немає. Важливим є видалення гематоми відкритим чи стерео–таксичним методом з урахуванням її обсягу, локалізації та впливу на структури мозку. Розроблено оригінальний метод стереотаксичного видалення нетравматичних внутрішньомозкових гематом. Проводяться дослідження з інтравентрикулярного введення тромболітиків при ГІ для прискорення санації шлуночкової системи, тобто в лікуванні мозкових розладів значно зросла роль нейрохірургії. Основу консервативного лікування ГІ становлять: 1) оптимізація артеріального тиску; загальні заходи щодо підтримки гомеостазу; 2) корекція супутніх неврологічних порушень; 3) заходи щодо профілактики й лікування таких соматичних ускладнень, як флеботромбоз, тромбоемболія легеневої артерії, пневмонія, гострі стресорні виразки травного тракту тощо [5, 7].
Подібними є лікувальні заходи при субарахноїдальному крововиливі: оптимізація рівня артеріального тиску за допомогою гіпотензивних препаратів, профілактика й лікування судинного спазму (блокатори кальцієвих каналів — німодипін), профілактика судом, протинабрякова терапія при розвитку набряку мозку, хірургічне лікування аневризм (кліпування шийки аневризми, балонна оклюзія тощо), гострої оклюзійної гідроцефалії, застосування гіпотермії [5]. Однак метод хірургічної декомпресії сьогодні залишається найбільш ефективним, він вірогідно підвищує виживання порівняно з пацієнтами, які отримували консервативну терапію.
Завдяки тому, що за три десятиліття після введення поняття ішемічної напівтіні уявлення про патогенез інсульту значно змінилося, у лікувальній тактиці відбувається перегляд пріоритетів: від концепції відновлення мозкового кровотоку в ішемізованих тканинах до концепції нейрорепарації, нейропротекції, що враховує пластичність мозку.
У зв’язку з цим дослідники наполягають на тому, щоб розглядати інсульт як цілком виліковне захворювання, а не як безвихідну ситуацію «неврологічної катастрофи». Тим більше що проведення реперфузії в межах терапевтичного вікна дозволило розширити можливості порятунку нейронів, які ще не зазнали не–оборотних змін. Саме у цьому напрямку сьогодні здійснюються численні науково-практичні розробки [28].
З огляду на те, що вже існують препарати для впливу на пенумбру, постало питання про засоби доставки такого препарату до місця дії. Такою речовиною, на думку багатьох дослідників, можуть бути ліпосоми. Ліпосоми можуть транспортувати ліки, надовго утримуючи їх у кровотоку, полегшуючи процес їх проникнення через гематоенцефалічний бар’єр у паренхіму мозку. На ліпосомах можна закріплювати будь-який нейропротектор, наприклад цитиколін, що добре зарекомендував себе, а також інші речовини, що знижують метаболізм мозку й по–повнюють енергетичні втрати.
Отже, нові перспективні підходи до нейропротекції, спрямованої на пато–генетичні ланки розвитку патологічного стану, відновлення діяльності нейронів, що вижили, а також ефективний пошук шляхів доставки нейропротекторів можуть допомогти в швидкому відновленні ішемізованої тканини головного мозку.
Сучасний стан і проблеми нейропротекторної терапії тяжкої черепно-мозкової травми та мозкового інсульту в Україні
Сьогодні для лікування постгіпоксичної енцефалопатії в Україні є великий вибір нейротропних препаратів, що мають антигіпоксантну дію [1, 4, 5, 28, 44].
Нейроцитопротектори — група препаратів, здатних підвищувати виживання нейронів в умовах гострої гіпоксії та ішемії, хоча єдиного саногенетичного механізму дії цієї групи лікарських засобів не існує. Тому підвищення ефективності кожного лікарського препарату можливе лише при раціональному використанні, тобто при застосуванні з урахуванням його фармакокінетики і фармакодинаміки та в оптимальних дозуваннях [28].
Ноотропи (наприклад, пірацетам) — речовини, які активують вищу інтегративну діяльність мозку, відновлюють порушені мнестичні й розумові функції, знижують неврологічний дефіцит і підвищують резистентність організму до екстремальних впливів. Але для реалізації ефекту ноотропів, що стимулюють енергетичний метаболізм, потрібна певна структурна цілісність тканини мозку і відповідний рівень функціональної активності мозку. Тому застосовувати пірацетам необхідно тільки при глибині порушення свідомості не нижче за 9–10 балів за шкалою коми Глазго. Більше того, ноотропні препарати викликають порушення ЦНС і протипоказані при порушеннях свідомості глибше за оглушення-сопор, тому що при будь-якому коматозному стані застосування засобів, що збуджують ЦНС, призводить до розгальмовування підкіркових структур, викликає психомоторне збудження або судоми, ще більше пригнічуючи кору. Отже, питання про використання ноотропів у хворих з тяжкою ЧМТ і МІ, ускладненими коматозним станом, є спірним, але вкрай актуальним. Доцільність використання в ранньому періоді гострої локальної ішемії мозку «пасток» вільних радикалів і препаратів, що руйнують перекиси (із сульфідними й тіоловими групами: унітіолу, тіосульфату натрію тощо), була доведена ще у 80-х роках минулого століття. Тоді ж було патогенетично обґрунтовано одночасне застосування токоферолів і каротиноїдів, що зв’язують каталізатори й інактивують синглетний кисень. Поряд з тіотриазоліном до сучасних антиоксидантів належить мексидол.
Мексидол (сіль 2-етил-6-метил-3-оксипіридину, структурного аналога вітаміну B6, і янтарної кислоти) є анти–оксидантом нового покоління. Він інгібує процеси перекисного окиснення ліпідів, активує ендогенну антиоксидантну систему супероксиддисмутази й церулоплазмін, запобігає зниженню активності глутатіон-залежних ферментів, унаслідок чого вірогідно зменшується активність процесів оксидантного стресу. Мексидол модулює активність мембранозв’язаних ферментів, рецепторних комплексів і посилює їхню здатність зв’язування з лігандами, що сприяє збереженню структурно-функціональної організації біомембран, транспорту нейромедіаторів і поліпшенню синаптичної передачі. Він має здатність посилювати компенсаторну активацію аеробного гліколізу, знижувати ступінь пригнічення окисних процесів у циклі Кребса в умовах гіпоксії зі збільшенням вмісту АТФ і креатинфосфату, активувати енергосинтезуючі функції міто–хондрій.
Одним з найбільш перспективних напрямів метаболічного захисту мозку від ішемії вважається безпосередній вплив на системи нейротрансмітерів і нейромодуляторів мозку, на нейрональні рецептори, створення умов для нормалізації співвідношення процесів збуджуючої і гальмівної нейротрасмісії [27]. Встановлено, що в основі когнітивних порушень при церебральній недостатності лежить холінергічна недостатність, яка зумовлена зниженням вироблення ацетилхоліну, порушенням балансу холінергічних ензимів, втратою холінергічних нейронів. Корекція холінергічної недостатності — важливий напрямок лікування когнітивних порушень при гострій церебральній недостатності різного генезу [28].
На сьогодні відомі два класи медикаментозних препаратів, застосування яких спрямоване на подолання холінергічної недостатності: 1) препарати, які безпосередньо поповнюють дефіцит ацетилхоліну, до них належать холіноміметик центральної дії — холіну альфосцерат (гліатилін), що є прекурсором ацетилхоліну, який проникає через гематоенцефалічний бар’єр, і цитидин-5-дифосфохолін натрію (цитиколін), який є донором холіну в процесах синтезу ацетилхоліну; 2) інгібітори ацетилхолінестерази: прозерин, галантамін, нейромідин тощо.
Цитиколін є незамінним попередником фосфатидилхоліну (лецитину), основного фосфоліпіду всіх клітинних мембран, включно з нейрональними мембранами, і є донором холіну в процесах синтезу ацетилхоліну. Цитиколін не виявляє прямої енерготропної дії. Препарат має виражений мітопротективний ефект. Як показано, цитиколін може зберігати цілісність внутрішньої мембрани мітохондрії, про що свідчить відновлення її потенціалу. Подібний механізм пов’язаний з відновленням рівня кардіоліпіну у внутрішній мембрані мітохондрій. Крім цього, виявлено, що цитиколін опосередковано, через збільшення активності глутатіон-зв’язаних ферментів (глутатіонредуктаза і глутатіонтрансфераза), регулює рівень відновленого глутатіону. Відновлений глутатіон, особливо мітохондріальний, гальмує окисну деструкцію Red-Oxi-чутливих ділянок мітохондріальної мембрани і формування стійкої мітохондріальної дисфункції. Показано, що через підвищення рівня відновленого глутатіону цитиколін може знижувати реакції нітрозуючого стресу і гальмувати NO-залежні механізми нейроапоптозу. Метааналіз результатів досліджень виявив ефективність цитиколіну при ІІ та ЧМТ, клінічні дослідження його ефективності тривають [28]. При ЧМТ цитиколін слід комбінувати з препаратами, що знижують ступінь нейронального й дифузного аксонального ушкодження. Отримано дані про ефективне поєднання цитиколіну з іншими нейропротекторами [1, 5, 27].
Гліатилін — холіноміметик з переважним впливом на ЦНС. При потраплянні в організм розщеплюється під дією ферментів на холін і гліцерофосфат: холін бере участь у біосинтезі ацетилхоліну, а гліцерофосфат є попередником фосфоліпідів нейрональної мембрани [28].
В ангіоневрології та нейротравматології застосовується низка препаратів (церебролізин, цереброкурин, кортексин, семакс), що впливають на нейропластичні, нейромедіаторні, нейропротективні, нейротрофічні й інтегративні процеси в мозку [28]. Реорганізація нейрональних процесів являє собою сукупність низки механізмів, що включають функціонування раніше неактивних шляхів, нейротрофічне відновлення волокон клітин, що збереглися, з формуванням нових синапсів, активацію нейрональних ланцюгів. Більше того, сьогодні все більшого значення набуває репаративна терапія, спрямована на поліпшення пластичності здорової тканини, яка оточує інфаркт мозку, активацію утворення полісинаптичних зв’язків, збільшення щільності рецепторів [27, 28].
Новим напрямом у дослідженні нейропептидів стало визначення їхньої ролі в регуляції нейроапоптозу, а також їхнього впливу на експресію генів раннього реагування. Нейропептиди вільно проникають через гематоенцефалічний бар’єр і справляють багатобічний вплив на ЦНС, що супроводжується високою ефективністю та вираженою спрямованістю дії за умови дуже малої їх концентрації в організмі. Багато нейропептидів мають виражені нейротрофічні ростові властивості, а також виявляють здатність регулювати експресію ранніх генів [44]. Відкриття нейротрофічних пептидних факторів спонукало до формування нової стратегії фармакотерапії — пептидергічної, або нейротрофної, терапії нейродегенеративних патологій.
Одним з перших препаратів нейропептидної природи є кортексин, який містить комплекс нейропептидів і являє собою ліофілізат, отриманий з мозку телят. Виражену ноотропну й церебропротективну активність продемонстрував нейропептидний препарат семакс АКТГ (4–10), що є гептапептидом (Met-Glu-His-Phe-Pro-Gly-Pro) [28].
Високоефективним препаратом, що сприяє активації церебральних і мультиорганних реституційних механізмів, є актовегін. Макро- і мікроелементи, що входять до складу актовегіну, є частиною нейропептидів, ферментів і аміно–кислот, тому значно краще засвоюються, розпізнаючись нейронами, ніж макро- і мікроелементи, що знаходяться у складі солей.
Церебролізин — препарат, що складається з низькомолекулярних нейропептидів (75 %) і вільних амінокислот (25 %) і отримується за стандартизованими біотехнологічними методиками з використанням ферментативного розщеплення очищених білків головного мозку свиней. Нейротрофічна активність церебролізину, подібна до активності природних нейротрофічних факторів, наприклад фактора росту нервів, була підтверджена низкою експериментальних досліджень. Один з механізмів антиапоптозної активності даного препарату пов’язаний зі зменшенням пошкоджуючої дії глутамату на нейрони.
Цереброкурин є одним з найбільш перспективних препаратів нейротрофічного ряду, який містить вільні амінокислоти, нейропептиди й низькомолекулярні продукти контрольованого протеолізу низькомолекулярних білків і пептидів ембріонів великої рогатої худоби. За механізмом дії і точками застосування цереброкурин принципово відрізняється від інших препаратів нейропептидної природи, зокрема від церебролізину. Цереброкурин містить пептиди, що несуть у собі програму аналізу стану й будівництва ЦНС. Отже, кінцевий ефект відрізняється через якісно відмінний механізм дії [27, 28]. В умовах гострої церебральної ішемії цереброкурин сприяв нормалізації в клітинах головного мозку функціональної активності мітохондрій, що забезпечувало ефективність мітохондріального окисного фосфорилювання й відновлення синтезу АТФ, блокування розвитку лактат-ацидозу й утворення вільних радикалів. Мітоспрямована нейро–протективна дія цереброкурину підтвердилася при встановленні його здатності впливати на ультраструктуру мітохондрій нейронів сенсомоторної кори та СА1-гіпокампа при експериментальному ІІ, ГІ і ЧМТ. Цереброкурин відновлював активність ферментів антиоксидантної та тіол-ди–сульфідної систем, що посилювало інактивацію АФК і додатково сприяло стабілізації окисних процесів клітин головного мозку в гострий період ішемії. У комплексі всі ці механізми протекторної дії забезпечували відновлення функціональної активності нейронів і гліальних клітин, а також синтез РНК у постішемізованих ділянках головного мозку.
Перспективним напрямом у лікуванні гострої церебральної недостатності є пошук ефективної комбінації лікарських засобів, що впливають на різні етапи патобіохімічних процесів і стимулюють відновлювальні процеси в нервовій тканині [1–5, 7–14]. Складність вирішення питання про поєднання тих чи інших нейротропних препаратів при лікуванні гострої церебральної недостатності різної етіології пов’язана з відсутністю даних щодо їх ефективності з позицій доказової медицини.
Взаємодія лікарських засобів відбувається за законами фармакодинаміки, фармакокінетики й фармакогенетики, що важливо враховувати при прогнозуванні результату взаємодії. Призначення лікарських препаратів і їх дози необхідно планувати для того, щоб виключити можливі побічні явища. В оптимальному варіанті краще використовувати три- або чотирикомпонентні комбінації лікарських речовин [28].
З огляду на багатокомпонентність пост–ішемічного каскаду було розроблено саногенетично виправдані комбінації лікарських препаратів, що потенціюють дію один одного при лікуванні ішемії будь-якої етіології. Ці комбінації дозволяють значно зменшити постішемічне нейрональне ушкодження, оскільки комплекс препаратів впливає практично на всі етапи ішемічного каскаду. У даному випадку не йдеться про поліпрагмазію, оскільки мішенню для кожного з препаратів є окрема ланка порочного кола постішемічного каскаду. Кожна з цих ланок не є слабкою, і перервати цей часто необоротний патогенетичний каскад можна саме із застосуванням комплексного впливу. Хоча адекватність дозових поєднань з урахуванням індивідуальної чутливості до них, прогноз можливих побічних ефектів та оцінка ступеня їхньої значущості в процесі лікування (співвідношення «ризик — користь») вимагають подальшого вивчення на більшій вибірці хворих.
Обговорюючи причини неефективності нейропротекції при гострій церебральній недостатності, багато авторів схиляються до думки про невідповідність експериментальних моделей. Відносно здорові експериментальні тварини, у яких інсульт викликається одномоментною оклюзією артерії, не можуть бути еквівалентні реальним пацієнтам, які часто страждають ще й від атеро–склерозу, цукрового діабету, артеріальної гіпертензії, ішемічної хвороби серця та інших хронічних захворювань, а також, як правило, мають низку факторів ризику (похилий вік, стать, паління тощо) [27, 28, 37, 39]. Тобто велике значення має індивідуальна сприйнятливість препаратів кожним конкретним пацієнтом. Низка авторів вважають, що, імовірно, проблема полягає в тому, що дослідники концентрують увагу на захисті нейронів. Однак інсульт — це не лише ушкодження й загибель нейронів, це хвороба головного мозку загалом. Дуже важливо звернути увагу на зміни, які відбуваються під час ішемії не лише в сірій, але й у білій речовині головного мозку. Недостатньо уваги приділяється також проблемі ушкодження мікро–циркуляторного русла і дещо умовної нейроваскулярної одиниці, що становить єдиний структурно-функціональний елемент тканини головного мозку. Очевидно, реалізація такого підходу на практиці означатиме необхідність підбору не одного або двох нейропротективних препаратів, а цілої схеми нейропротективного лікування.
Нині низкою дослідників і клініцистів порушується питання доцільності комбінованого призначення препаратів первинної і вторинної нейропротекції з метою відновлення кровотоку й мета–болізму нервових клітин, а також регуляції процесів клітинної загибелі [1]. Основною причиною загибелі нейронів у зоні ішемічної півтіні є глутаматний каскад, а також оксидативний стрес. У каскаді глутаматної та оксидативної нейротоксичності одним з етапів, що призводять до загибелі нейронів, є зміна характеру експресії генів — індукторів апоптозу і генів — інгібіторів апоптозу, порушення окисно-відновних процесів за рахунок блокади мітохондріального комплексу [3].
У лікуванні ІІ, ГІ та ЧМТ важливе місце належить засобам вторинної нейро–протекції (антиоксиданти, метаболітотропні препарати, нейропептиди, ноотропи).
Членом-кореспондентом НАМН України, професором В.І. Чернієм розроблена комбінована й експериментально апробована поетапна нейропротекція. Первинна нейропротекція (перші 4 доби після експериментального ІІ та ЧМТ) включала L-лізину есцинат, латрен, тіотриазолін, династат, венофундин. Вторинна нейропротекція (21 доба після ІІ та ЧМТ) включала Тіоцетам, актовегін, цераксон, цитофлавін, цереброкурин. Встановлено, що поетапне комбінування засобів первинної і вторинної нейропротекції за розробленою схемою безпосередньо або опосередковано здатне модулювати експресію генів раннього реагування c-fos і, отже, запускати програму синтезу адаптаційних білків (у тому числі HSP і фактора, індукованого гіпоксією (HIF)) у нейронах в умовах гострої церебральної ішемії та значно підвищувати їхню концентрацію в головному мозку. Встановлено зростання концентрації в тканинах головного мозку HSP і HIF-1, які за рахунок шаперонної активності, стабілізації актинових філаментів перешкоджають розвитку некрозу. Максимальне модулювання експресії генів раннього реагування c-fos мало місце у тварин, яким після експериментальної ішемії проводилося комплексне лікування, що включає первинну і вторинну нейропротекцію.
Експериментально на моделі ІІ та ЧМТ доведено, що патофізіологічний підхід до двохетапного нейропротективного впливу здатний посилювати експресію гена c-fos, змінювати морфологічний тип загибелі нейронів, переключаючись на більш м’який апоптичний шлях, який є оптимальним, впорядкованим процесом припинення життєдіяльності деструктивно змінених нейронів.
Отже, реалізація нейропротективної дії препаратів первинної та вторинної нейропротекції відбувається, мабуть, за рахунок їхньої здатності підвищувати концентрацію в тканинах головного мозку HSP-білків. В умовах ішемічного пошкодження головного мозку білки HSP і HIF перешкоджають розвитку некрозу за рахунок позитивного впливу на синтез антиоксидантних ферментів, шаперонної активності, стабілізації актинових філаментів. Крім того, низкою робіт була показана роль підвищення експресії HSP70 у клітинах мозку (в астроцитах) у захисті їх від загибелі, викликаної кисневим голодуванням. Також було продемонстровано здатність очищеного препарату HSP70 підвищувати виживання нейронів, які беруть участь у глутаматергічній синаптичній передачі в нюховій корі мозку щурів, –після руйнівного впливу тяжкої аноксії [58]. Беручи до уваги дані про здатність HSP посилювати життє–здатність нейрональної клітини в умовах гіпоксії та факт взаємодії HSP і HIF, що відіграє першорядну роль у клітинній відповіді на гіпоксію, можна припустити, що HSP бере участь у регуляції сигнальних шляхів відповіді клітини на гіпоксичний стрес на рівні регуляції стабільності HIF.
/12_u.jpg)
Нейропротективна дія поетапної нейро–протекції проявлялася в збільшенні щільності гліальних клітин і нейронів у корі головного мозку як на 4-ту, так і на 21-шу добу експерименту, а також підвищенні їх морфофункціональної та транскрипційної активності (збільшення вмісту РНК) і зниженні кількості деструктивно змінених клітин. Максимальний рівень щільності та площі клітин нейронів, концентрації РНК зафіксовано на 21-шу добу експерименту в 4-й серії монгольських піщанок, які отримували первинну і вторинну нейропротективну терапію. У нейронах гіпокампа максимальна кількість гена c-fos (гена первинної або ранньої відповіді) і білка Bcl-2 (білок-супресор апоптозу (програмованої клітинної загибелі)) також виявлена на 21-шу добу експерименту в 4-й серії монгольських піщанок, які отримували первинну і вторинну нейропротективну терапію. Інтегральний показник нейропротективної дії препаратів як первинної, так і вторинної протекції — це зменшення неврологічного дефіциту, про що свідчило вірогідне зниження середнього бала за шкалою C.P. McGrаw, а їхня комбінація мала більш виражений вплив на досліджуваний показник. Саме в цій 4-й серії експериментальних тварин на 21-шу добу виявлено максимальне зменшення неврологічного дефіциту — вірогідне зниження середнього бала за шкалою C.P. McGraw. У тварин, яким –після на–несення експериментальної стандартизованої травми проводилося комплексне лікування, що включає лізин, реосорбілакт, гекодез, тіотриазолін, латрен, династат, Тіоцетам, актовегін, цераксон, цереброкурин, спостерігалася значна –активація компенсаторно-пристосувальних проце–сів, зокрема пролі–ферація нейроглії, –нейрональний сателітоз, низький ступінь мікрогліозу в поєднанні зі зниженням щільності нейронів з ознаками некрозу й апоптозу [4–14, 28].
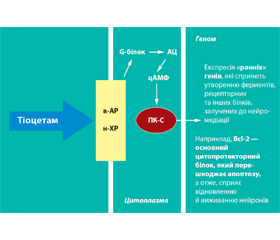
Сьогодні на фармацевтичному ринку України є величезна кількість препаратів з нейропротекторними властивостями, але немає можливості проведення мультицентрових рандомізованих плацебо-конт–рольованих досліджень цих нейропротекторів. При гострій мозковій ішемії, викликаній ЧМТ або МІ, кожен хворий є певним конкретним завданням, що потребує вирішення, зі своїми особ–ливостями й проблемами. Як варіант, є перспективним застосування нового церебропротективного препарату, який є комбінацією пірацетаму з тіотриазоліном (Тіоцетам). Не виключено, що він може і повинен застосовуватись у всіх періодах перебігу ЧМТ, але ефект може бути дозозалежним.
Тіоцетам — це препарат, який поєднує ноотропні, мнемотропні, антигіпоксичні властивості пірацетаму з протиішемічними, антиоксидантними й мембраностабілізуючими властивостями тіотриазоліну, чим він вигідно відрізняється від пірацетаму та інших аналогічних ноотропів.
Найбільшу активність має комбінація, що складається з 0,05 г тіотриазоліну і 0,2 г пірацетаму. На ґрунті цієї комбінації була розроблена лікарська форма — таблетки й ампули Тіоцетам. Комбінація продемонструвала широкий спектр церебропротективних і ноотропних ефектів. Про це свідчить ціла низка експериментальних досліджень, проведених у Запорізькому державному медичному університеті. Експериментальними дослідженнями доведено, що фармакологічний ефект Тіоцетаму обумовлюється взаємопотенціюючою дією тіотриазоліну і пірацетаму. Комплексний препарат Тіоцетам, розроблений українськими вченими, збільшує інтенсивність функціонування метаболічного ГАМК-шунта і концентрацію ГАМК в ішемізованих тканинах, нормалізує нейромедіаторний (дофамін- і норадренергічний) обмін, знижує вираженість вестибулярних розладів, покращує асоціативні процеси, інтегративну діяльність мозку, стимулює процеси мислення і пам’яті, покращує здатність до концентрації та навчання.
Він прискорює утилізацію глюкози при аеробному й анаеробному шляху окиснення, збільшує фонд АТФ, здатний активувати компенсаторні цитозольно-мітохондріальні шунти енергії в умовах гострої ішемії, знижує ультраструктурні порушення мітохондрій, гальмує реакції оксидативного стресу в тканинах мозку, підвищує рівень відновлених інтермедіатів тіол-дисульфідної системи, підвищує активність мітохондріальної супероксиддисмутази (СОД), гальмує нейроапоптоз [39]. Тіоцетам чинить сприятливу дію на навчання і пам’ять тварин після експериментальних ЧМТ, ІІ та ГІ, введення кетаміну або барбітуратів. Тіоцетам справляє модулюючий вплив на експресію генів раннього реагування c-fos, підвищує мРНК HIF-1α у нейронах СА1-гіпокампа і сенсомоторної кори головного мозку щурів з експериментальною патологією при навчанні в радіальному лабіринті. Тіо–цетам знижує кількість помилок робочої та просторової пам’яті, збільшує збереження набутої навички.
В інтенсивній терапії гострої церебральної недостатності, обумовленої тяжкою черепно-мозковою травмою або мозковим інсультом, актуальною залишається проблема визначення термінів початку лікування Тіоцетамом і критеріїв вибору оптимальної дози Тіоцетаму за допомогою методу інтегрального кількісного аналізу ЕЕГ-патернів для вивчення реактивності мозку. Членом-кореспондентом НАМН України, професором В.І. Чернієм розроблено методику визначення адекватної дози Тіоцетаму на основі зіставлення методу оцінки порушення свідомості за шкалою коми Глазго і методу інтегрального кількісного аналізу — рівня дезорганізації ЕЕГ-патерну з дослідженням реактивності мозку у відповідь на введення Тіоцетаму. Обстежено 235 (112 чоловіків і 123 жінки) постраждалих віком від 17 до 72 років з тяжкою ізольованою черепно-мозковою травмою. Клінічно розрізняли такі види пошкодження головного мозку при тяжкій і вкрай тяжкій ізольованій ЧМТ: забій головного мозку тяжкого ступеня, внутрішньо–черепні гематоми: шкала коми Глазго — 4–8 балів. Протягом 7 діб після терапії з включенням до стандартного протоколу препарату Тіо–цетам були виявлені найнижчі (порівняно з групою контро–лю, яка отримувала терапію за стандартним протоколом) рівні систолодіастолічного співвідношення (S/D) та індексу пульсації в хребетних артеріях, що свідчило про значне зниження циркуляторного опору у вертебробазилярному басейні. Починаючи з першої-другої доби після травми досліджено реактивність мозку на введення Тіоцетаму. Вводять 5 мл препарату і за зміною електроенцефалограми, записаної до початку введення препарату і через 20–30 хвилин після закінчення введення Тіоцетаму, визначають тип реакції ЦНС. Якщо типи реакцій на введення свідчать про збільшення дезорганізації патерну, то введення препарату припиняють, але на наступний день дослідження повторюють, і роблять так до моменту появи дезорганізації ЕЕГ-патерну. Після цього повертаються до попередньої дози, її залишають. Тестують на наступний день на адекватній дозі (не викликає посилення дезорганізації) і так далі. Паралельно проводять оцінку порушення свідомості за шкалою коми Глазго і кореляційний аналіз між застосованою дозою Тіоцетаму і рівнем свідомості за шкалою коми Глазго (бали), кількісними показниками ЕЕГ (інтегральні показники). У результаті буде рекомендована доза препарату залежно тільки від результатів дослідження і рівня свідомості за шкалою коми Глазго. Такий підхід дозволить з великим ступенем чутливості судити про дію Тіоцетаму при тій чи іншій мірі порушення свідомості (оглушення-сопор-кома). Вихідною дозою для введення Тіоцетаму у пацієнтів з тяжкою ЧМТ при рівні неврологічного дефіциту до 10 балів за шкалою коми Глазго є 5 мл (0,5 г). Під контролем нейрофізіологічного (ЕЕГ, ультразвукова транскраніальна допплерографія) і клінічного моніторингу добова доза Тіоцетаму може бути поступово (крок 5 мл) збільшена до 20 мл максимально [28].
Ефективність Тіоцетаму в умовах подібних морфофункціональних змін ЦНС пов’язана, імовірно, зі здатністю препарату, як і інших рацетамів, різноспрямовано впливати на основні нейромедіаторні системи в різних ділянках головного мозку. Він збільшує вміст і оборот серотоніну у фронтальній корі мозку і зменшує в смугастому тілі, гіпоталамусі й стовбурі мозку, підвищує рівень і оборот дофаміну у фронтальній корі й гіпоталамусі та знижує в смугастому тілі. Особливо цікавими є можливості Тіоцетаму посилювати синтез і викид ацетилхоліну, зворотне захоплення холіну в мозку і підвищувати чутливість і число мускаринових рецепторів, що особливо важливо в умовах функціональної недостатності таламокортикальних взаємодій і висхідних активуючих холінергічних впливів.
Доведено, що в гострому періоді ЧМТ Тіоцетам спричиняє ослаблення процесів нейрональної загибелі та зниження частоти цитолізу в сенсомоторній зоні фронтальної кори та СА1 зоні гіпокампу, що, крім інших протиішемічних дій (інтенсифікація аеробних шляхів окиснення глюкози, підвищення фонду макроергічних фосфатів, активація дихального ланцюга мітохондрій і посилення сателітозу гліальних клітин), може забезпечувати фармакологічний ефект при подібних морфофункціональних змінах ЦНС.
У результаті досліджень члена-кореспондента НАМН України, професора В.І. Чернія [28] встановлено:
1. Нейропротективна дія Тіоцетаму (антиоксидантна, протиішемічна, –антиамнестична) вигідно відрізняється від дії пірацетаму та інших препаратів рацетамного ряду і похідних ГАМК.
2. В інтенсивній терапії гострої церебральної недостатності, зумовленої тяжкою черепно-мозковою травмою або мозковим інсультом, актуальною залишається проблема визначення термінів початку лікування Тіоцетамом і критеріїв вибору оптимальної дози препарату.
3. Вибір початкової дози Тіоцетаму в пацієнтів із ЧМТ і ГПМК має бути диференційованим і проводитися під нейро–фізіологічним контролем.
4. Включення препарату до комплексу інтенсивної терапії в гострій фазі захворювання передбачає проведення моніторингу неврологічного статусу, дослідження спектральної потужності ЕЕГ, вивчення реактивності ЦНС у відповідь на фармакологічний вплив.
У КНП «9-та міська клінічна лікарня» м. Запоріжжя було обстежено 79 хворих з легкою і середньотяжкою закритою бойовою черепно-мозковою травмою (13–14–15 балів за шкалою коми Глазго). Вік хворих — від 23 до 55 років. У більшості випадків (76) ЧМТ спричиняла мінно-вибухова травма. Клінічно більшість ЧМТ представлена струсом головного мозку — 67, у 12 був забій головного мозку легкого ступеня, у 2 випадках діагностовано переломи склепіння черепа. Усі хворі надходили до нейрохірургічного відділення в гострому періоді (1–3 доби з моменту поранення). Слід відзначити, що в 100 % випадків ЧМТ поєднувалася з акубаротравмою.
У дослідженнях використано клінічні й функціональні методи оцінки стану центральної нервової системи: клінічне й неврологічне обстеження хворого; електроенцефалографія; метод систематизації, класифікації і кодування електроенцефалограм людини; топографічне картування електроенцефалограми з розрахунком спектральної потужності й когерентності; метод інтегрального кількісного аналізу ЕЕГ; метод нейромережевого моделювання; ультразвукове допплерографічне дослідження судин мозку. Інтра–церебральні заходи, що проводились у 1, 2, 3-му періодах травматичної хвороби головного мозку: аналгоседація; мембраностабілізація і первинна нейропротекція; усунення церебрального вазоспазму; поліпшення венозного відтоку з порожнини черепа; профілактика й лікування набряку і набухання головного мозку; відновлення енергетичного дефіциту нервової тканини та вторинна нейропротекція; відновлення нейромедіаторного обміну та вторинна нейропротекція. Інтрацеребральні заходи, що проводились у 4-му періоді травматичної хвороби головного мозку: нейрореабілітація, нейромедіаторна і нейро–трофічна терапія.
У клінічній частині цієї роботи аналізуються результати комплексного лікування, включно із застосуванням Тіоцетаму, проведеного 39 хворим з легкою та середньотяжкою закритою бойовою ЧМТ. З метою контролю взято групу із 40 хворих, у яких застосовувалась стандартна терапія. Підбір хворих в основній і контрольній групах відбувався з урахуванням тяжкості ЧМТ, віку і супутньої патології. Тіоцетам вводився протягом 5 днів внутрішньовенно краплинно з першої доби надходження пацієнтів у відділення нейрореанімації в середній дозі 0,2–0,3 мл/кг на 150 мл фізіологічного розчину протягом години. У першу чергу для оцінки нейропротективної дії комбінованої терапії з включенням до неї розчину для парентерального застосування Тіоцетам у гострому періоді ЧМТ як специфічний маркер у сироватці крові пацієнтів визначалася нейрон-специфічна єнолаза (NSE). Також у сироватці крові визначався білок S100, який належить до класу нейромаркерів і відображає активність нейроглії як закономірну відповідь на масивне руйнування нейронів. Отже, вивільнення ферменту й білка з нейронів у кров є маркером ушкодження нервової тканини. Активність NSE (нг/мл) і вміст S100 (нг/мл) визначали методом твердофазного імуноферментного аналізу з використанням наборів NSE ELISA kit #ab217778 (ABCAM, USA) і S-100 ELISA kit #LS-F6208 (LifeSpan Biosciences Inc., USA). Як маркер оксидативного стресу і для оцінки антиоксидантної дії досліджуваної комплексної терапії з включенням Тіоцетаму в сироватці визначали нітротирозин методом твердофазного імуноферментного аналізу з використанням наборів Nitrotyrosine ELISA kit #HK501 (Hycult Biotech, USA). Дослідження проводили на мікропланшетному імуноферментному рідері Sirio-S (seac Radim Company, Італія).
Результати дослідження розраховували із застосуванням стандартного статистичного пакета Statistica® for Windows 6.0 (StatSoftInc., № AXXR712D833214FAN5), а також SPSS 16.0, Microsoft Office Excel 2003. Нормальність розподілу оцінювали за критерієм Shapiro-Wilk. Дані були подані у вигляді середнього значення. Віро–гідність різниці між середніми значеннями визначали за критерієм Стьюдента (у випадку нормального розподілу). У випадку розподілу, відмінного від нормального, або аналізу порядкових змінних використовували критерій U Mann-Whitney. Для порівняння незалежних змінних у більше ніж двох вибірках використовували дисперсійний аналіз (ANOVA) при нормальному розподілі або критерій Kruskal-Wallis для розподілу, відмінного від нормального. Для всіх видів аналізу статистично значущою вважалась різниця р < 0,05 (95 %).
Наші дані вказують на порівняно ранній регрес суб’єктивних симптомів струсу головного мозку та забою середнього ступеня в постраждалих від закритої бойової ЧМТ основної групи порівняно з пацієнтами контрольної групи. Так, після 5 діб у 9 % відзначався періодичний помірний головний біль, у той час як у контрольній групі він відзначався у 20 %. Помірно виражені астенічні й когнітивні порушення після 5 діб спостерігалися у 9,7–14,3 % пацієнтів досліджуваної групи, у конт–рольній групі — у 18–27 %. При цьому раннє застосування Тіоцетаму в досліджуваних пацієнтів починаючи з другої доби не викликало підвищення збудливості, нервозності, порушення сну, тобто симптомів, характерних для прийому великих доз рацетамних ноотропів (пірацетам, фенотропіл, прамістар, оксирацетам, аніпацетам тощо).
При дослідженні вегетативної реактивності після проведеного лікування препаратом Тіоцетам порівняно з конт–рольною групою було виявлено, що показник нормальної вегетативної реактивності зріс до 60 %, тоді як у контрольній групі — до 42 %. Зменшилися показники пацієнтів з підвищеною вегетативною реакцією — з 42,8 до 17,9 %, у контрольній групі вони знизилися з 38,5 до 23,1 %. Позитивний клінічний ефект в основній групі пацієнтів, які приймали Тіоцетам, був відзначений у перші 5 днів у 94 % пацієнтів. У контрольній групі позитивний клінічний ефект був відзначений у 76 % спостережень. Поліпшення спостерігалося як у клінічній сфері, так і за даними показників вегетативної реактивності ЕЕГ, шкали дослідження когнітивних функцій MMSE. Побічних реакцій та ознак непереносимості препарату ми не спостерігали в жодного пацієнта протягом усього періоду спостереження в клініці.
Лабораторними дослідженнями було встановлено, що в крові в пацієнтів з легкою і середньотяжкою закритою бойовою черепно-мозковою травмою (13–14–15 балів за шкалою коми Глазго) у гострий період активність маркера мембранної цілісності нейронів NSE була вірогідно підвищена щодо відносно здорових, що вказує на розвиток суттєвої деструкції нейронів і превалювання некротичного типу їх загибелі. Рівень іншого маркера, білка S-100, що відображає активність астроцитарної глії, зміна якої є закономірною відповіддю нервової тканини на некротичні й некробіотичні процеси, значно зріс відносно значень умовно здорових людей. Отримані дані свідчать про первинне ураження масиву нейронів після травми. Також у крові пацієнтів з легкою і середньотяжкою закритою бойовою черепно-мозковою травмою зросла концентрація нітротирозину, що свідчить про активацію оксидативного стресу. Включення в базисну терапію ЧМТ Тіоцетаму приводило до зниження рівня нейромаркера S-100 на 44,7 % щодо показників пацієнтів при надходженні (p < 0,05). Базисна терапія без Тіоцетаму приводила до зниження S-100 на 26,8 % (p < 0,05). Включення в базисну терапію ЧМТ –Тіоцетаму приводило до зниження нейромаркера NSE на 31,3 % щодо показників пацієнтів при надходженні (p < 0,05). Базисна терапія без Тіоцетаму приводила до зниження NSE на 25 % (p < 0,05). Курсове призначення Тіоцетаму у складі базової терапії ЧМТ призводить до зниження маркера оксидативного стресу нітротирозину на 53,7 % щодо групи пацієнтів при надходженні (p < 0,05). Базисна терапія ЧМТ без Тіоцетаму знижує нітротирозин на 22,5 % (p < 0,05). Отримані дані свідчать про підвищення нейропротективної та антиоксидантної дії засобів базової терапії при включенні до неї Тіоцетаму.
Висновки
Застосування Тіоцетаму (5 днів, в/в, 0,2–0,3 мл/кг на 150 мл фізіологічного розчину) в комплексному лікуванні легкої та середньотяжкої закритої бойової ЧМТ (усього 79 хворих) приводило до поліпшення як у клінічній сфері (у 94 % пацієнтів — позитивний ефект, а в контролі — 76 %), так і за даними показників вегетативної реактивності ЕЕГ, шкали дослідження когнітивних функцій MMSE. Включення Тіоцетаму в комплексне лікування ЧМТ призводило до зниження нейромаркерів: S-100 — на 44,7 % і NSE — на 31,3 %, а також маркера оксидативного стресу — нітротирозину на 53,7 % (p < 0,05). Значення молекулярних маркерів у групі, що отримувала Тіо–цетам, вірогідно відрізнялися від показників контрольної групи. Отже, проведене комплексне клінічне й нейрофізіологічне дослідження Тіоцетаму показало доцільність і патогенетичну обґрунтованість його застосування в гострому періоді легкої і середньотяжкої закритої черепно-мозкової травми, у тому числі бойової (13–14–15 балів за шкалою коми Глазго). Застосування Тіоцетаму в гострому періоді легкої та середньотяжкої закритої бойової черепно-мозкової травми підвищує нейропротективну дію базисної терапії, гальмує оксидативний стрес, сприяє ранньому усуненню неврологічного дефіциту й відновленню когнітивних функцій. Отримані результати клінічно обґрунтовують перспективність включення Тіоцетаму як нейропротектора до базисної терапії закритої бойової ЧМТ. Поєднання ноотропного і нейропротекторного ефекту, закладеного в оригінальному комбінованому складі препарату Тіо–цетам (пірацетам + тіотриазолін), дозволяє уникнути побічних дій, характерних для традиційних рацетамних ноотропів, за рахунок зниження дози пірацетаму і потенціюючої дії тіотриазоліну.
Список литературы
Список літератури знаходиться в редакції