
Журнал «Здоровье ребенка» 3 (24) 2010
Вернуться к номеру
Молекулярно-генетические механизмы развития и современные методы лечения легочной артериальной гипертензии у детей 2. Молекулярные медиаторы развития легочной артериальной гипертензии
Авторы: Волосовец А.П., Национальный медицинский университет им. А.А. Богомольца, г. Киев; Абатуров А.Е., Петренко Л.Л., Кривуша Е.Л., Днепропетровская государственная медицинская академия
Рубрики: Педиатрия/Неонатология
Версия для печати
В статье рассмотрены современные представления о патогенетических молекулярных механизмах развития легочной артериальной гипертензии, в основе которых лежат наследственно обусловленные или приобретенные нарушения функционирования генов, участвующих в поддержании сосудистого тонуса, ангиогенезе, формировании экстрацеллюлярного матрикса. Рассмотренные патогенетические механизмы формирования легочной артериальной гипертензии позволяют отнести ее в группу ангиопролиферативных болезней, требующих разработки новых целевых лекарственных средств, способствующих восстановлению микроциркуляторного кровотока в легочной ткани и направленных не только на вазодилатацию, но и на ингибицию пролиферации.
Легочная артериальная гипертензия, молекулярно-генетические механизмы, дети.
Сокращения: АКМ — активные кислородсодержащие метаболиты, АМФ — аденозинмонофосфат, ГМФ — гуанозинмонофосфат, ЛАГ — легочная артериальная гипертензия, ИЛАГ — идиопатическая ЛАГ, СЛАГ — семейная ЛАГ, 5HTR — серотониновый рецептор, Angt — ангиотензин, ANP — предсердный натрийуретический пептид, ArgRS — аргинин-тРНК синтаза, BMP — костные морфогенетические белки, BMPR2 — рецептор II типа костного морфогенетического белка, CD36 — рецептор тромбоспондина, cIAP-1 — baculoviral IAP repeat-containing protein 1, COX — циклооксигеназа, DAG — диацилглицерол, EDCF — эндотелиновый контрактильный фактор, EDHF — эндотелиновый фактор гиперполяризации, EDNR — эндотелиновый рецептор, ET — эндотелин, FGF2 — фактор роста фибробластов 2, Flt-1 — fms-связанная тирозинкиназа 1, H 2 O 2 — перекись водорода, HIF — индуцибельный гипоксией фактор, ICAM-1/CD54 — межклеточная адгезивная молекула 1, ICAM-2/CD102 — межклеточная адгезивная молекула 2, IFN — интерферон, IP-10/CXCL10 — интерферон- g -индуцируемый протеин 10 kDa, KDR — kinase insert domain receptor, MIP-1 a /CCL3 — макрофагальный воспалительный протеин 1 a , MIP-1 b /CCL4 — макрофагальный воспалительный протеин-1 b , MIG/CXCL9 — монокин, индуцирующий g -интерферон, MLCK — киназа легких цепей миозина, MMP — матриксная металлопротеиназа, NF-1 — нуклеарный фактор 1, NFAT — нуклеарный фактор активации Т-лимфоцитов, NF-IL6 — нуклеарный фактор IL6, NF- k В — нуклеарный фактор k В, NO — монооксид азота, NOS — нитрооксидсинтаза, PAI-1 — ингибитор активатора плазминогена 1, PlGF — фактор роста плаценты, PDGF — тромбоцитарный фактор роста, PECAM-1/CD31 — platelet endothelial cell adhesion molecule-1, PGI 2 — простациклин, PGIS — простациклинсинтаза, PKC — протеинкиназа C, PL A — фосфолипаза A, PL C — фосфолипаза С, PL D — фосфолипаза D, PPAR — активируемые пероксисомальным пролифератором (мероксизонпролифератор-активированные) рецепторы, PpET — препроэндотелин, РrоЕТ-1 — проэндотелин-1, PSGL-1 — Р-селектиновый гликопротеинный лиганд 1, RANTES/CCL5 — регулятор активации нормальной Т-клеточной экспрессии и секреции, SERT — серотониновый транспортер, TGF- b — трансформирующий фактор роста b , TIMP-1 — тканевый ингибитор металлопротеиназ, Tph1 — триптофангидроксилаза 1, TX A2 — тромбоксан A2, TXAS — тромбоксансинтаза, VCAM-1 — адгезивная молекула 1 сосудистого эндотелия, VEGF — сосудистый эндотелиальный фактор роста, VEGFR-2 — рецептор 2 сосудистого эндотелиального фактора роста, VE-кадгерин — эндотелиальный кадгерин, VIP — вазоактивный интестинальный пептид, vWF — фактор Виллебранда.
Молекулярные компоненты патогенеза ЛАГ
Развитие ЛАГ сопровождается значительными изменениями экспрессии множества генов. Исследование экспрессии 6800 генов легочной ткани показало, что у больных с ИЛАГ и СЛАГ наблюдается усиление экспрессии 133 и ингибиция экспрессии 174 генов [81].
Основные молекулярные структуры, определяющие развитие ЛАГ, представлены в табл. 1.
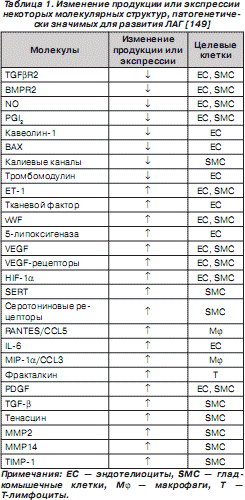
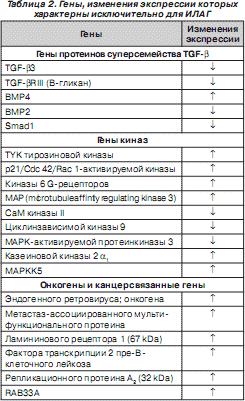
Характерно, что изменение экспрессии некоторых генов — некдин-связанного протеина, индуцибельного гена B 94 протеина тумор-некротизирующего фактора, ДНК последовательности клона 322 B 1 хромосомы 22 q 11–12, метиониновой аминопептидазы, CO-029 опухоль-ассоциированного антигена, триггера 1 транспортабельного элемента, кавеолина, субъединицы репликационного протеина A 70 kDa, ArgRS, P311 HUM (3.1), аполипопротеина E, ингибитора моноцитарно-нейтрофильной эластазы, гена тромбомодулина, BMP3b, кальциневрина A2, p126 (ST5), фактора транскрипции цинкового пальца hEZF (EZF) — наблюдается при любых формах ЛАГ. Но изменения экспрессии генов рецептора III TGF- b , BMP2, митогенактивируемой протеинкиназы киназы 5 (MAPKK5), RACK 1, аполипопротеина C - III , ламининового рецептора 1 характерны исключительно для пациентов с ИЛАГ (табл. 2) [81].
Эндотелий-ассоциированные медиаторы, участвующие в развитии ЛАГ
Эндотелиоциты принимают непосредственное участие в регуляции тонуса и роста сосудов, артериального давления, обеспечении оптимального уровня свертываемости крови, проницаемости стенок сосудов, защиты организма от инфекционных агентов, продуцируя в определенных условиях множество таких биологически активных веществ, как вазоконстрикторы (Ang t II, простагландин Н 2 , TX A2 , супероксидный анион, EDCF , ET ), вазодилататоры (аденозин, NO , H 2 O 2 , PGI 2 , EDHF, С-натрийуретический пептид) факторы коагуляции (Ang t -IV, PAI-1, тромбоцитактивирующий фактор, PDGF, тромбопластин, vWF, ET -1), антитромботические факторы (антитромбин, гепарин, NO , PGI 2 , тканевый активатор плазминогена, тромбомодулин), продукты экстрацеллюлярного матрикса (ламинин, коллаген, протеогликаны, протеазы, фибронектин), факторы, обеспечивающие защиту организма (хемокины, адгезивные молекулы, интерфероны, провоспалительные цитокины), факторы роста (инсулиноподобные факторы роста, FGF2, TGF , VEGF) [79, 101, 218, 219].
В условиях физиологической нормы экспрессия генов эндотелиоцитов достаточно стабильная. Внешние или внутренние причинно-значимые разрешающие факторы, изменяющие условия функционирования или повреждающие эндотелий легочных сосудов, обусловливают изменение спектра биологически активных веществ, продуцируемых эндотелиоцитами. Активация эндотелиоцитов может быть связана с влиянием гипоксии, гемодинамических факторов, патоген-ассоциированных молекулярных структур (РАМР) инфекционных агентов, провоспалительных цитокинов (IL-1 b , TNF- a , IFN- g ) и др. Активация эндотелиоцитов обусловливает вазоконстрикцию, повышение коагуляционного потенциала крови, пролиферацию эндотелиоцитов [23, 57, 80, 147, 216].
Монооксид азота
Монооксид азота, продуцируемый эндотелиоцитами, выполняет определенную роль в регуляции тонуса легочных сосудов. Эндотелиальная NOS ( eNOS ), катализируя конверсию L -аргинина в цитруллин, производит NO , который активирует гуанилатциклазу, что приводит к повышению внутриклеточной концентрации цГМФ в гладкомышечных клетках, вызывая их релаксацию [23, 144]. При ЛАГ наблюдается снижение уровня NO в стенках легочных артерий, несмотря на то что экспрессия eNOS может быть даже повышенной. Считают, что внутриклеточно произведенный NO не высвобождается из клетки во внеклеточное пространство, а захватывается аппаратом Гольджи [180].
Нарушение баланса продукции простаноидов
Простаноиды — простагландины ( PGE 2 , PGI 2 , PGD 2 , PGF 2 a ) и тромбоксаны являются дериватами арахидоновой кислоты и оказывают свое действие, связываясь со специфическими G -протеиновыми рецепторами [8].
Простаноиды активно участвуют в регуляции апоптоза, миграции клеток и модуляции синтеза разнообразных молекул эндотелиоцитами (табл. 3).
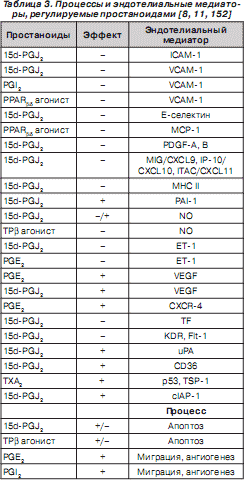
Развитие ЛАГ сопровождается нарушением баланса продукции различных простаноидов за счет снижения синтеза вазодилататора и ингибитора пролиферации гладкомышечных клеток PGI 2 , при усилении продукции вазоконстриктора и индуктора пролиферации гладкомышечных клеток TX A 2 [163].
Эндотелиоциты отличаются высоким уровнем содержания COX -1 и PGIS, а тромбоциты — COX -1 и TXAS, в связи с чем при активации PL A 2 преобладающим метаболитом арахидоновой кислоты у эндотелиоцитов является PGI 2 , а у тромбоцитов — TX A 2 [169]. Активация эндотелиоцитов сопровождается ингибицией продукции PGI 2 и увеличением продукции TX A 2 [8].
PGI 2 , образующийся из простагландина H 2 под действием PGIS, является одним из важнейших эндогенных вазодилататоров, действие которого связано с его способностью стимулировать образование цАМФ. PGI 2 также ингибирует агрегацию тромбоцитов и пролиферацию гладкомышечных клеток сосудов [160, 208, 209]. PGI 2 вызывает физиологические эффекты, взаимодействуя с двумя видами G-протеиновых рецепторов — мембранными рецепторами PGI 2 (IP), которые активируют аденилатциклазу, и внутриклеточными рецепторами PPAR b / d , ингибирующими пролиферацию клеток [97, 142, 170]. У больных с ЛАГ наблюдается дефицитарная экспрессия рецепторов PGI 2 и ингибиция активности PGIS в эндотелиоцитах мелких и средних легочных артерий [177].
При ЛАГ наблюдается не только усиленная продукция как эндотелиоцитами, так и тромбоцитами одного из самых мощных вазоконстрикторов — TX A 2 , но и увеличение экспрессии его рецепторов (TP) на эндотелиоцитах, тромбоцитах, гладкомышечных клетках сосудов и клетках правого желудочка сердца [56, 181].
Эндотелин-1
В настоящее время показано, что легочная гипертензия ассоциирована с высокой продукцией ET-1 эндотелиоцитами легочных сосудов [21, 64]. Эндотелиальный фактор констрикции, выделенный M. Yanagisawa и соавт. [3] в 1980 году, был идентифицирован в 1988 году и получил название эндотелина. В настоящее время эндотелиновое семейство, принадлежащее к суперсемейству неприлизина, представлено 4 гомологичными по химической структуре эндотелинами — ЕТ-1, ЕТ-2, ЕТ-3 и ЕТ-4. Молекулы всех ET состоят из 21 аминокислотного остатка. Однако, несмотря на высокую гомологичность, гены различных ET расположены на разных хромосомах: ген ЕТ-1 расположен на хромосоме 6 (6p24.1), ген ЕТ-2 — на хромосоме 1, а ген ЕТ-3 — на хромосоме 20 (20q13.2–q13.3) [47, 110]. В ткани легкого самые высокие концентрации характерны для ЕТ-1. Факторами, стимулирующими продукцию ЕТ-1, являются: 1) механическая стимуляция эндотелия; 2) гипокапния; 3) влияние биологически вазоактивных веществ (тромбина, ионов Ca 2+ , адреналина, Ang t II, вазопрессина, допамина, эритропоэтина); цитокинов (TNF- a , IL-l, IL-l b , IL-6); факторов роста (TGF- b , FGF2, инсулиноподобных факторов роста); липидов (липопротеидов низкой и высокой плотности). Субстанциями, ингибирующими синтез ET, являются NO , цГМФ, ANP, PGI 2 , брадикинин [85, 174, 187]. Продуценты ЕТ-1 — эндотелиоциты, эпителиоциты, гладкомышечные клетки, тканевые макрофаги, кардиомиоциты, нейроны [174]. Под воздействием индукторов эндотелиногенеза в клетках первоначально синтезируется PpET, продукция которого регулируется факторами транскрипции — АР-1 (c-fos и c-jun), NF-1, GATA-2. PpET состоит из 203 аминокислотных остатков. В цитоплазме клетки PpET расщепляется специфическими эндопептидазами с образованием большого ЕТ-1 или РrоЕТ-1, состоящего из 38 аминокислотных остатков [119]. Расщепление эндотелинпревращающим ферментом молекулы РrоЕТ-1 в области Trp21 (Endothelin Converting Enzyme — ЕСЕ) Val22 [179] ведет к образованию ЕТ-1 [53]. В эндотелиоцитах ЕТ-1 сохраняется в удлиненных везикулах, известных как тельца Weilbel-Palade [215]. Концентрация ЕТ-1 в плазме крови здоровых людей составляет 0,7–5 пг в 1 мл. Элиминация ЕТ-1 из плазмы крови преимущественно осуществляется легкими, время полураспада ЕТ-1 составляет приблизительно 7 минут [154, 175].
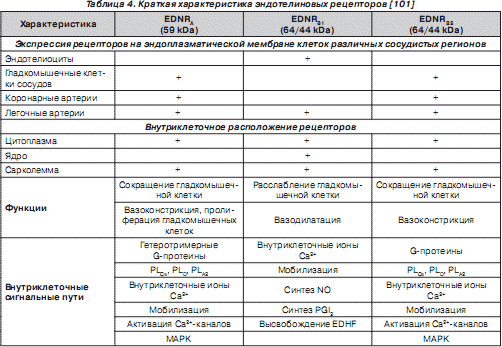
Эндотелины взаимодействуют с тремя типами эндотелиновых рецепторов — EDNR A , EDNR В , EDNR С . Рецепторы EDNR A связывают преимущественно ЕТ-1, а EDNR В — ЕТ-1, ЕТ-2 и ЕТ-3, EDNR С — ЕТ-3. EDNR A расположены в основном на гладкомышечных клетках сосудов, опосредуя вазоконстрикторный эффект ЕТ-1, и на гладкомышечных клетках респираторного тракта, кардиомиоцитах, гепатоцитах, нейронах, остеобластах, меланоцитах, адипоцитах и разнообразных клетках репродуктивной системы. EDNR В расположены на эндотелиоцитах, гладкомышечных клетках некоторых сосудов (аорты, брыжеечных артерий, коронарных артерий, вен), гепатоцитах, нейронах, нейроглиях, остеобластах [48, 101, 110, 119]. Гены данных рецепторов расположены на хромосомах 4 (4q31.23) и 13 (13q22) соответственно [110]. Экспрессия EDNR В максимально выражена на цитоплазматической мембране эндотелиальных клеток. EDNR В представлены двумя изоформами — EDNR В1 , участвующими в эндотелийзависимой вазодилатации, и EDNR В2 , обусловливающими вазоконстрикторный эффект (табл. 4) [63, 219].
ЕТ-1 также вызывает и бронхоконстрикцию, способствует развитию воспаления, усиливая хемотаксис, адгезию и активацию нейтрофилов. При легочной гипертензии снижается способность легких утилизировать ЕТ-1, что приводит к повышению его уровня в циркулирующей крови. ЕТ-1, активируя EDNR A , через G q/11 -белок индуцирует внутриклеточные фосфолипазы (PL Cβ , PL D , PL А2 ) и через G i -протеин ингибирует аденилатциклазу [62, 165]. Повышение активности PL C β приводит к образованию инозитол-1,3,5-трифосфата ( IP 3, IP 4) и DAG . IP 3, IP 4 увеличивают активность мембранных низковольтажных Са 2+ -каналов ( VOC ), способствуют высвобождению ионов Са 2+ из саркоплазматического ретикулума, обеспечивая повышение внутриклеточной концентрации Са 2+ , что приводит к сокращению миозина [60]. DAG активирует PKC , которая фосфорилирует киназу легких цепей миозина ( MLCK ), обусловливая сокращение миозина [101]. Однако ЕТ-1, активируя EDNR B 1 , индуцирует внутриклеточные сигнальные пути, которые обусловливают продукцию и вазодилатирующих факторов — NO , PGI 2 , EDHF [101]. У больных с ЛАГ гиперпродукция ЕТ-1 сопровождается резким уменьшением синтеза PGI 2 , NO и VIP [98, 100]. ЕТ-1 увеличивает экспрессию HIF -1 a , который активирует синтез VEGF [59]. Повышение активности PLА 2 усиливает метаболизм арахидоновой кислоты. Повышение уровня внутриклеточной концентрации Са 2+ приводит к активации COX-1, индуцируя продукцию EDCF [56, 217]. ЕТ-1 способствует активации фактора транскрипции NFAT , который, в свою очередь, индуцирует экспрессию антиапоптотического протеина Bcl -2, ингибирующего апоптоз эндотелиоцитов [207].
Серотонин
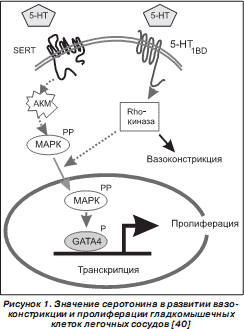
Развитие ЛАГ сопровождается повышением уровня концентрации серотонина (5-гидрокситриптамин — 5HT), который играет важнейшую роль в патогенезе данного заболевания [187]. Показано, что ЛАГ сопровождается усилением экспрессии в эндотелиоцитах гидроксилазы Tph1, которая участвует в синтезе серотонина из триптофана [120, 121]. Серотонин оказывает влияние на клетку, взаимодействуя с мембранными рецепторами 5HTR или попадая во внутриклеточное пространство при помощи SERT. В настоящее время показано, что в развитии легочной гипертензии из 14 идентифицированных 5HTR принимают участие 5HTR 1B , 5HTR 2A и 5HTR2B [120]. У пациентов с ИЛАГ наблюдается высокая частота встречаемости L-типа полиморфизма промотора гена SLC6A4 SERT (65–75 %), который сопровождается высокой экспрессией SERT в тромбоцитах и клетках гладких мышц легочных артерий. Предполагают, что LL-SERT генотип связан с ранней манифестацией легочной гипертензии [49, 189, 190–192]. Ген SLC6A4 расположен на длинном плече хромосомы 17 (17q11.1–q12) [120]. SERT сигналы ведут к серотонинзависимому ремоделированию сосудов и пролиферации сосудистых гладкомышечных клеток [188]. Препараты, подавляющие аппетит, синтезированные на основе фумарата, вызвали в 60-х годах прошлого века в Швейцарии резкое увеличение количества больных с ЛАГ [89]. В последующем было установлено, что производные фенфлурамина являются лигандами SERT [173]. Так, активация SERT сопровождается генерацией АКМ, которые фосфорилируют p42/44, ERK1/2 MAP киназы. Активация рецепторов 5-HT1B/D приводит к индукции Rho-киназы, без активации которой не происходит серотонин-индуцированная пролиферация гладкомышечных клеток. Активность Rho-киназы обусловливает констрикцию легочных сосудов, а также транслокацию в ядро клетки ERK, фосфориляцию фактора транскрипции GATA4, что индуцирует пролиферацию гладкомышечных клеток (рис. 1) [40, 133]. Серотонин ингибирует ВМР сигнальный путь Smad и экспрессию ВМР-индуцибельных генов [187]. Также серотонин ингибирует вольтаж-зависимые калиевые каналы Kv1.5, что приводит к активации Ca 2+ -каналов L-типа и повышению уровня внутриклеточной концентрации ионов Ca 2+ , обусловливая сокращение гладкомышечных клеток стенок легочных сосудов [120]. Серотонин стимулирует продукцию представителя семейства кальций-связывающих белков S100 протеина S100A4 (метастазина 1), который активирует MAPK ERK1/2 и фактор транскрипции GATA4, стимулируя миграцию и пролиферацию гладкомышечных клеток сосудов [163].
Фактор Виллебранда, тромбомодулин, молекулы адгезии
Практически при всех формах ЛАГ у больных наблюдается высокая концентрация vWF в сочетании со снижением экспрессии мембранного рецептора тромбина тромбомодулина и увеличенная продукция Р-селектина (маркера эндотелиальной дисфункции) [6].
Фактор vWF, связывая гликопротеидный рецептор тромбоцитов Ib/IX с субэндотелиальным коллагеном, фиксирует тромбоциты в местах повреждения эндотелия. Повышенная продукция vWF увеличивает коагуляционный потенциал крови и коррелирует с возникновением преждевременной смерти. Дефицит экспрессии тромбомодулина сопровождается недостаточностью синтеза антикоагуляционного белка С [23, 107, 124, 214].
Индукция экспрессии Р-селектина, который в состоянии покоя, как и vWF, сохраняется в тельцах Weibel-Palade, на поверхности мембраны эндотелиоцитов в результате его взаимодействия с лейкоцитарным PSGL-1 обеспечивает роллинг моноцитов, нейтрофилов, эффекторных Т-лимфоцитов, В-клеток, натуральных киллеров [115].
При ЛАГ наблюдается усиление экспрессии эндотелиальных адгезивных молекул ICAM-1/CD54, VCAM-1, которые взаимодействуют с лейкоцитарными интегринами (ICAM-1 — с b 2-интегрином, VCAM-1 — с a 4 b 1-интегрином/VLA-4) (табл. 5). Усиление экспрессии эндотелиоцитами молекул адгезии способствует рекрутированию лейкоцитарных популяций, их трансмиграции и развитию воспалительного процесса [6, 77]. Также при ЛАГ увеличивается экспрессия представителя иммуноглобулинового суперсемейства — адгезивной молекулы PECAM -1/CD31 — трансмембранного гликопротеина, имеющего 6 экстрацеллюлярных иммуноглобулиноподобных доменов. PECAM -1 является механосенситивной молекулой, которая в ассоциации с VE-кадгерином и VEGFR-2 реагирует на изменения кровотока в сосудистом русле [78].

Факторы роста VEGF, FGF2 и PDGF
В состоянии «покоя» эндотелиоциты не секретируют VEGF, FGF2. Однако после индукции наблюдается активная продукция данных факторов роста клетками эндотелия. При ЛАГ характерен высокий уровень экспрессии VEGF и FGF 2 [23, 51, 58].
Семейство VEGF состоит из пяти представителей: VEGF-A, VEGF-B, VEGF-C, VEGF-D и PlGF. VEGF-А наиболее активно экспрессирован в области плексиформных поражений, сопровождающих тяжелое течение ЛАГ. VEGF — основной митоген, фактор выживания и дифференцировки эндотелиальных клеток, а FGF2, наряду с серотонином, является одним из ведущих индукторов пролиферации гладкомышечных клеток [70].
Взаимодействие VEGF с рецептором VEGFR -2 активирует PL C , преимущественно PL C g и PL C b 3 . Фосфолипаза PL C g принимает участие в тубулогенезе, дифференцировке и синтезе ДНК, а PL C b 3 — в реорганизации актина, миграции и пролиферации клетки [44]. Возбуждение VEGFR -2 также приводит к активации факторов транскрипции STAT 1 и STAT 3, усилению продукции PGI 2 и повышению экспрессии eNOS в эндотелиоцитах [43, 102, 223]. В свою очередь, STAT3 усиливает транскрипцию генов VEGF. Наличие при ЛАГ положительной обратной связи между процессами активации STAT3 и продукции VEGF обусловливает их пролонгированную гиперэкспрессию. Длительная активность STAT3 сопровождается постоянной продукцией антиапоптотических протеинов, в частности Bcl -2, что обусловливает формирование апоптозрезистентных и гиперпролиферирующих эндотелиоцитов, играющих определяющую роль в образовании плексиформных поражений с тонкостенными расширениями [102]. Активация VEGFR -2 также инактивирует каспазу-9 и Bad [223]. Возбуждение VEGFR -2, активируя компоненты сигнальных внутриклеточных путей p38 MAPK и паксиллина, обусловливает миграцию эндотелиальных клеток [36]. Миграция клеток в субэндотелиальное пространство приводит к формированию слоя клеток и появлению экстрацеллюлярного матрикса между эндотелием и внутренней эластической пластиной. Данное морфологическое образование является патогномоничным для ЛАГ и получило название неоинтима [140].
FGF2 — представитель большого семейства гепарин-связывающих факторов роста, который синтезируется как эндотелиоцитами, так и макрофагами и фибробластами. Данный фактор роста проявляет свое пролиферативное действие через активацию специфического рецептора FGFR гладкомышечных клеток, что приводит к индукции PKC и, как следствие, к синтезу противоапоптотических протеинов — сурвивина, XIAP и Bcl-XL [58]. FGF2 также играет ключевую роль в ангиогенезе [69].
Протеины семейства PDGF (PDGF-A, PDGF-A, PDGF-С, PDGF- D ) действуют как мощные митогены и хемоаттрактанты на гладкомышечные клетки легочных сосудов и являются факторами роста для фибробластов и глиальных клеток [10, 150, 156]. Индуцируют экспрессию PDGF TGF- b , эстроген IL-1, FGF2, TNF- a и липополисахарид. PDGF-B преимущественно экспрессирован эндотелиоцитами, мегакариоцитами и нейронами, PDGF-A и PDGF-C — эпителиоцитами, мышечными клетками, предшественниками нейронов, PDGF-A — фибробластами [10]. ЛАГ сопровождается повышенным уровнем продукции PDGF и экспрессии его рецепторов PDGFR- a , PDGFR- b . Рецепторы PDGFR активируют внутриклеточные сигнальные пути Ras-MAPK, PI3K и PL C , стимулируя перестройку актиновых нитей и мобилизацию ионов Ca 2+ [151]. Применение иматиниба — антагониста PDGFR подавляет процесс ремоделирования сосудистой стенки при ЛАГ [177].
Моноамины
При ЛАГ в ткани легкого характерно увеличение концентрации естественных моноаминов — диаминов (путресцина, кадаверина), олигоаминов (спермидина, спермина), которые способны усиливать процессы сосудистого ремоделирования [94, 202].
Значение медиатора неадренергической, нехолинергической системы — вазоактивного интестинального пептида в развитии ЛАГ
В настоящее время многочисленными исследованиями показано, что представитель суперсемейства секретинов вазоактивный интестинальный пептид, как медиатор неадренергической, нехолинергической системы, играет ключевую физиологическую роль в вазодилатации легочных сосудов, ингибиции процессов пролиферации гладкомышечных клеток. VIP нейтрализует вазоконстрикторное действие гипоксии, ET, лейкотриена D 4, амилоидного b -пептида [177, 220]. Показано, что TGF - b и цилиарный нейротрофический фактор, действуя синергично, индуцируют синтез VIP . Активированный изоформами TGF - b комплекс Smad2,3/Smad4, транслоцируясь в ядро клетки, способен взаимодействовать с двумя различными сайтами цитокин-реагирующего элемента CyRE промотора гена VIP , усиливая его транскрипцию [130, 177]. VIP и питуитарный пептид, взаимодействуя с G-рецепторами VPAC 1 и VPAC 2 , которые высоко экспрессируются клетками ткани легкого, активируют аденилатциклазу, что ведет к индукции образования цАМФ, продукции NO [87]. В дополнение к сосудистым эффектам VIP оказывает противовоспалительные эффекты: 1) модулирует пролиферацию и активацию T-клеток; 2) ингибирует факторы транскрипции NF- k B, NFAT ; 3) ингибирует синтез провоспалительных цитокинов (TNF- a , IL-6, IL-12, IL-18), хемокинов (RANTES); 4) усиливает продукцию противовоспалительного IL-10. Показано, что у больных с ЛАГ наблюдается снижение продукции VIP , а мутации в интронах 1, 2, 3 и 4 (g.448G>A g.501C>T g.764T>C g.2267A>T g.2390C>T g.3144T>C g.3912A>G g.4857A>G) гена VIP могут привести как к повышению давления в легочных сосудах, так и к облитерации сосудов [90, 221].
Эндотелиально-мезенхимальный клеточный переход
Под влиянием разрешающих патогенетически значимых медиаторов — TGF - b , FGF 2 происходит модуляция экспрессии генов некоторых эндотелиоцитов, которая постепенно изменяет их фенотип. Первоначально в эндотелиоцитах ингибируется синтез E -кадгерина, b -катенина, Tie -2, тирозинкиназный рецептор III типа/Flk-1, других эндотелий-специфических протеинов и активируется синтез экстрацеллюлярных матриксных металлопротеиназ, что приводит к потере контакта данных клеток с окружающими эндотелиоцитами и их миграции в субэндотелиальное пространство. Последующая активация экспрессии ассоциированных с транслокацией протеинов Notch1 или Notch4 обусловливает переход эндотелиоцита в фибробласт или в гладкомышечно-подобную клетку, экспрессирующую a -актин [14].
Заключение
Современные представления о молекулярных механизмах развития легочной артериальной гипертензии позволяют не только верифицировать данную патологию как заболевание, которое обусловлено мутациями или приобретенными нарушениями функционирования генов BMPR2, ALK-1, эндоглина, SERT, VIP , а также, возможно, и других генов, участвующих в поддержании сосудистого тонуса, ангиогенезе, формировании экстрацеллюлярного матрикса, но и отнести ЛАГ к группе ангиопролиферативных болезней. По всей вероятности, назрела необходимость смены стратегии лечения ЛАГ с парадигмы «вазодилатация» на «вазодилатация и ингибиция пролиферации». Разработка новых целевых лекарственных средств, которые будут ингибировать основные патогенетические молекулярные структуры, вызывающие вазоконстрикцию, пролиферацию, миграцию эндотелиоцитов, гладкомышечных клеток, синтез белков экстрацеллюлярного матрикса, и которые будут способствовать восстановлению микроциркуляторного кровотока в легочной ткани, позволит надеяться, что в недалеком будущем медицина достигнет реальных успехов в лечении ЛАГ у детей, которые будут измеряться не пятилетней выживаемостью, а качеством жизни.
Список литературы находится в редакции