
Журнал «Здоровье ребенка» 6 (41) 2012
Вернуться к номеру
Болезнь Вильсона — Коновалова
Авторы: Сенаторова А.С., Омельченко Е.В., Урываева М.К.
Харьковский национальный медицинский университет, кафедра педиатрии № 1 и неонатологии
Ермолаев М.Н., Чуб Е.И., Пушкарь Е.М.
Харьковская областная детская клиническая больница
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
В статье приведены данные об этиологии, патогенезе и клинике болезни Вильсона — Коновалова, а также описан сложный случай собственного клинического наблюдения болезни Вильсона — Коновалова (гепатолентикулярная дегенерация) у девочки 9 лет.
У статті наведені дані про етіологію, патогенез та клініку хвороби Вільсона — Коновалова, а також описано складний випадок особистого клінічного спостереження хвороби Вільсона — Коновалова (гепатолентикулярної дегенерації) у дівчинки 9 років.
Data on etiology, patogenesis, and clinical picture of Wilson — Konovalov disease were considered in the article, and also there is described difficult clinical case of own clinical observation of Wilson — Konovalov (hepatolenticular degeneration) disease in 9-year-old girl.
болезнь Вильсона — Коновалова, манифестация, диагностика, лечение.
хвороба Вільсона — Коновалова, лікування, діагностика, маніфестація.
Wilson — Konovalov diasease, diagnostics, children.
Болезнь Вильсона — Коновалова (БВК) — наследственное заболевание, передающееся по аутосомнорецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтранcпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона — Коновалова встречается во всех регионах мира с частотой около 30 случаев на 1 млн человек. В регионах, где существуют близкородственные браки, частота возрастает. Гетерозиготные носители составляют примерно 1 % клинически здоровых людей; хотя заболевание у них не развивается, биохимическое исследование выявляет субклинические нарушения метаболизма меди. Болезнь Вильсона — Коновалова должна предполагаться у каждого индивидуума в возрасте от 3 до 45 лет с патологией печени неустановленной этиологии [1, 3, 11].
Первооткрыватель заболевания — А.К. Вильсон, описавший болезнь в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3q21.1).
Организм человека содержит около 50–100 мг меди. Суточная потребность в меди для человека — 1–2 мг; 95 % абсорбированной в кишечнике меди транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5 % — в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохромСоксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтеза главного медьсвязывающего белка (церулоплазмина) и выведения меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению. При быстром высвобождении меди возникает ферментопенический гемолиз [2, 7].
При наличии БВК с рождения клинические симптомы до 5летнего возраста возникают крайне редко. В типичных случаях БВК манифестирует в подростковом и юношеском возрасте. Основные методы диагностики БВК представлены ниже [4, 6, 9]:
1. Снижение церулоплазмина сыворотки < 0,2 г/л.
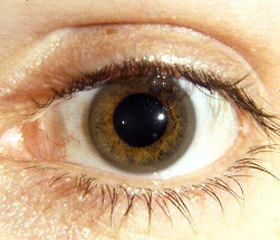
2. Обнаружение кольца Кайзера — Флейшера на роговице пациентов.
3. Повышение суточной экскреции меди с мочой > 100 мкг.
4. Повышение концентрации меди в ткани печени > 250 мкг/г сухой массы при норме 25 мкг/г.
5. Генетическая диагностика у сибсов, позволяющая выявить наличие у пациента генетических мутаций.
6. Компьютерная или магнитнорезонансная томография головного мозга позволяет визуализировать гиподенсивные очаги в базальных ганглиях и лентикулярном ядре.
Клинические проявления зависят от соотношения аккумуляции меди в тех или иных органах.
Выделяют пять форм болезни Вильсона — Коновалова: брюшная, ранняя, дрожательноригидная, дрожательная, экстрапирамиднокорковая [5, 8, 10].
При брюшной форме отмечается тяжелое поражение печени, приводящее к гибели больного раньше появления признаков со стороны нервной системы. Заболевают этой формой чаще дети.
Ранняя форма начинается в возрасте от 7 до 15 лет и отличается быстрым течением. При этом преобладает скованность движений, насильственные движения. Лицо имеет застывший в гримасе вид. Речь нечеткая, характерны судорожные смех и плач, судорожные припадки, взрывы раздражения, некоторое снижение интеллекта.
Дрожательноригидная форма начинается в юношеском возрасте и встречается чаще других. Протекает она несколько медленнее остальных форм, с ремиссиями (состояниями вне обострения) и ухудшениями. Появляется одновременно скованность движений и дрожание, усиливающееся при напряжении мышц, движениях и волнении. Дрожание захватывает конечности, туловище и голову.
Дрожательная форма начинается в возрасте 20–30 лет, протекает медленно, среди признаков заболевания преобладает дрожание.
Экстрапирамиднокорковая форма встречается реже других форм, но протекает тяжело. На первый план выходят признаки поражения головного мозга с судорожными припадками, парезами (временным обездвиживанием отдельных участков тела) и тяжелыми нарушениями интеллекта.
Приводим собственное наблюдение.
Девочка А., 9 лет, поступила в Областную детскую клиническую больницу с жалобами на отечность лица, туловища, нижних конечностей, увеличение в размерах живота, боли в коленных и голеностопных суставах, боли в животе, повышение температуры тела до фебрильных цифр, геморрагическую сыпь на коже лица, нарушение сна, снижение аппетита.
Из анамнеза болезни известно, что за год до появления жалоб ребенку проводилась экстракция зуба, через 6 месяцев повысилась температура тела до субфебрильных цифр, появились боли в суставах, периодически рвота, увеличение в размерах живота. В связи с присоединением выраженного отечного синдрома ребенок госпитализирован в ЦРБ с диагнозом «острый гломерулонефрит». Последний был исключен. В связи с повышением уровня печеночных ферментов (АЛТ — до 13 норм, АСТ — до 12 норм) заподозрен острый вирусный гепатит. Девочка госпитализирована в Областную детскую клиническую инфекционную больницу (ОДКИБ).
При поступлении в ОДКИБ общее состояние ребенка было расценено как тяжелое за счет печеночной недостаточности (в биохимическом анализе крови выявлено повышение уровня общего билирубина в два раза за счет прямого, значительный цитолиз, снижение уровня общего белка до 49 г/л — норма 65–72 г/л). При определении маркеров хронических гепатитов были обнаружены IgG к HBcor. При проведении компьютерной томографии органов брюшной полости выявлены признаки цирротической трансформации печени. Установлен диагноз «Хронический гепатит В, тяжелая форма. Цирроз печени. Печеночная энцефалопатия. Асцит». Проводилась инфузионная терапия глюкозосолевыми растворами, антибиотиками, диуретиками, гепатопротекторами. Состояние ребенка не улучшалось, проявления асцита нарастали. Для проведения лапароцентеза переведена в хирургический стационар, где ежедневно проводился забор асцитической жидкости, продукция которой составляла до 1,7–1,9 л/сут. В стационаре перелита свежезамороженная плазма, осуществлялась дезинтоксикационная и симтоматическая терапия. На 7е сутки, после уменьшения явлений асцита, снова переведена в инфекционную больницу. Несмотря на наличие дренажа в брюшной полости, мать ребенка категорически отказалась от проводимого лечения и забрала ребенка на 8е сутки пребывания в стационаре.
Спустя три дня девочка из центральной районной больницы вновь госпитализирована в тяжелом состоянии в отделение реанимации Областной детской инфекционной больницы. При обследовании: анемия — 90 г/л, цитолиз: АЛТ — до 9 норм, АСТ — до 7 норм, гипербилирубинемия — до 61,2 мкмоль/л за счет прямой фракции, обнаружены антинуклеарные антитела и антитела к нативной и денатурированной ДНК, гипопротеинемия — до 46,7 г/л, гипергаммаглобулинемия — 51,6 %. При эзофагогастродуоденоскопии: эритематозная гастродуоденопатия, варикозное расширение вен пищевода I–II степени, признаки портальной гипертензии. Заподозрен аутоиммунный гепатит. Для дальнейшего лечения была переведена в гастроэнтерологическое отделение Областной детской клинической больницы.
Из анамнеза жизни известно, что ребенок родился от I беременности, протекавшей на фоне фетоплацентарной недостаточности. Роды стремительные в сроке гестации 39–40 недель. Родилась в асфиксии. Оценка по шкале Апгар — 5 баллов. Росла и развивалась нормально. Аллергологический анамнез: пищевая аллергия с 6месячного возраста. Семейный анамнез: у отца — хронический алкогольный гепатит.
В отделении состояние ребенка оставалось тяжелым. Через неделю после поступления в клинику отмечалась эвентрация пряди сальника в лапароцентезное отверстие, по поводу чего проведена резекция пряди с ушиванием эвентрационного отверстия в хирургическом стационаре. При объективном обследовании обращали на себя внимание проявления астенизации. Кожа бледная, на коже лица — геморрагическая сыпь, пастозность век, суставы нормальных размеров и формы, отеки на нижних конечностях. Со стороны органов дыхания — без особенностей. Со стороны сердечнососудистой системы — границы относительной сердечной тупости в пределах нормы, тоны сердца звучные, тахикардия, выслушивался систолический шум в 4м межреберье слева от грудины. Живот увеличен в размерах. Печень и селезенка не пальпировались. Диурез снижен.
Клинический анализ крови: гемоглобин — 72 г/л, эритроциты — 2,58 ´ 1012/л, тромбоциты — 90 ´ 109/л, лейкоциты — 13,4 ´ 109/л, гранулоциты — 71,4 %, лимфоциты — 26,5 %, моноциты — 2,1 %, СОЭ — 4 мм/ч.
В биохимическом анализе крови: общий белок — 42 г/л (норма 65–85 г/л), аминотрансферазы: АЛТ — до 9,5 ммоль/ч/л (норма до 0,14 ммоль/ч/л), АСТ — 5,1 ммоль/ч/л (норма до 0,14 ммоль/ч/л), холестерин — 1,2 ммоль/л (норма 3,15—5,17 ммоль/л), bлипопротеиды — 7 усл.ед. (норма 35–55 усл.ед.), общий билирубин — 48,1 мкмоль/л, прямой билирубин — 27,5 мкмоль/л, непрямой — 20,6 мкмоль/л. Уровень антител к нативной ДНК — 50 ед. (норма 25 ед.), денатурированной ДНК — 45 ед. (норма 25 ед.), антинуклеарных антител — 2,4 ед. (норма 1,1 ед). Ревматоидный фактор — 40 МЕ/мл (норма до 20 МЕ/мл), антистрептолизин О — 400 ед. (норма до 250 МЕ/мл).
УЗИ органов брюшной полости: печень: размеры увеличены (правая доля — 15,6 ´ 12,4 ´ 8,6 см, левая доля — 9,6 ´ 8,4 ´ 6,5 см), края утолщены, капсула уплотнена. Умеренный диффузный фиброз, нерезкий холангит. Воротная вена расширена до 10 мм; желчный пузырь: 45 ´ 16 мм, отек стенки до 6 мм, ложе желчного пузыря интенсивно фиброзировано; поджелудочная железа: увеличена (головка — 22 мм, тело — 16 мм, хвост — 1,9 мм), структура неоднородная, эхогенность повышенная, перидуктальный фиброз; селезенка: увеличена (13,8 ´ 9,2 ´ 7,3 см), капсула четкая, структура однородная, периваскулярный фиброз, селезеночная вена в области ворот селезенки расширена (до 11,8 см); почки: правая 119 ´ 60 мм, левая 117 ´ 58 мм, увеличены, паренхима утолщена до 20 мм, гиперэхогенна, контрастность пирамидок, пиелоэктазии нет.
При компьютерной томографии органов брюшной полости и забрюшинного пространства, проведенной в спиральном режиме шагом 5 мм: печень увеличена в размерах однородной структуры, пониженой плотности до 45–50 мм, портальная вена диаметром 13 мм. Селезенка в размерах не увеличена, 119 ´ 38 мм, гомогенной структуры, плотностью 42–44 мм. Поджелудочная железа не изменена. Почки несколько увеличены, чашечнолоханочная система не изменена, определяются мезентериальные лимфатические узлы диаметром 5–7 мм, парааортальные лимфатические узлы диаметром до 4–5 мм. Мочевой пузырь не изменен. Гиперплазии тазовых лимфатических узлов не выявлено. Костнодеструктивных изменений позвонков костей таза не выявлено. Заключение: данные в пользу диффузного поражения печени.
При компьютерной томографии плечевых суставов, проведенной в спиральном режиме шагом 3 мм, при мультипланарной реконструкции деструктивных изменений головок плечевых костей не выявлено, метадиафизы плечевых костей не изменены, кортикальный слой сохранен, левая и правая лопатки без патологических изменений. Мягкие ткани верхнего плечевого пояса — без особенностей.
Заподозрена болезнь Вильсона — Коновалова, в связи с чем ребенку проводилось 3кратное исследование показателей церулоплазмина и меди сыворотки крови, уровень экскреции меди с мочой. Выявлено снижение церулоплазмина сыворотки крови — до 153, 98, 72,8 мг/л (при норме 200–540 мг/л). Отмечался также низкий уровень меди крови в динамике — 9,5, 11,4, 8,5 мкМ/л (при норме 12–24 мкМ/л). Однако суточная экскреция меди с мочой оставалась в пределах нормы — 41,0, 52,6, 60,2 мкг/сут (при норме 1,5–70,0 мкг/сут). Генетическое консультирование болезнь Вильсона — Коновалова поставило под сомнение.
Консультация гематолога: аутоиммунная тромбоцитопеническая пурпура, острое течение, средней степени тяжести.
Консультация окулиста: патологии со стороны органов зрения не выявлено, кольцо Кайзера — Флейшера не обнаружено.
Во время лечения в гастроэнтерологическом отделении (в течение 6 недель) состояние ребенка оставалось тяжелым. Самочувствие постепенно улучшалось — девочка стала более активной, аппетит — удовлетворительным. При ходьбе сохранялась утиная походка, которую ребенок связывал с болями в голеностопных суставах.
Периодически предъявляла жалобы на боли в грудной клетке, шумное дыхание при нарастании чувства голода, однократно — носовое кровотечение. На фоне повышения температуры тела до 38 °С отмечался болевой синдром в коленных, плечевых, голеностопных суставах, левой половине грудной клетки, ребрах. Девочка отказывалась вставать с кровати, принимать пищу. Появилась многократная рвота. Кожные покровы оставались бледными. На коже лица и в области шеи мелкая геморрагическая сыпь. Обращал на себя внимание стойкий отечный синдром.
Учитывая жалобы, клиническую симптоматику (прогрессирующая печеночная недостаточность с синдромами нарушения белковосинтетической, печеночноклеточной функции печени, вторичные нарушения гемостаза, выраженный отечный синдром, а также аутоиммунный компонент в развитии заболевания у ребенка: суставной синдром в виде артралгий, тромбоцитопения иммунного генеза, наличие антител к нативным и денатурированным белкам, антинуклеарных антител, значительное повышение уровня гаммаглобулинов и острофазовых показателей), а также проведенную дифференциальную диагностику, ребенку был установлен диагноз: «Аутоиммунный гепатит, тип I, с высокой степенью активности, осложненный портальной гипертензией; печеночная недостаточность 1й степени. Аутоиммунная тромбоцитопеническая пурпура, острое течение, средней степени тяжести. Хронический вирусный гепатит В, неактивный (фаза интеграции)».
Назначено лечение: постельный режим, диета — стол № 5, глюкокортикоиды: преднизолон 1,5 мг/кг/сут в течение одной недели с повышением дозы до 2,0 мг/кг/сут в течение трех недель, в последующем доза преднизолона была разделена на равные введения перорально и в/в струйно, с постепенным снижением на 5 мг в неделю. Для коррекции водноэлектролитных нарушений, стабилизации онкотического давления, купирования отечного синдрома в терапии назначались инфузии свежезамороженной плазмы по 200 мл № 8, инфузии 10% альбумина по 200 мл № 4 — еженедельно), поляризующей смеси (10% глюкоза, инсулин, 7,5% хлорид калия); диуретические препараты в/в: фуросемид 1,0, верошпирон 3 мг/кг/сут с постепенным снижением до 1 мг/кг; гепатопротекторы в/в струйно — гептрал по 400 мг № 20, гепасол по 200 мл № 10; препараты урсодезоксихолевой кислоты — урсохол 20 мг/кг/сут; антибиотик — цефуроксим 750 мг 3 раза в сутки; противогрибковые препараты: флуконазол 100 мг 1 раз в сутки; симптоматическая и метаболическая терапия (лактулоза 15,0 мл 4 раза в день, смекта, аспаркам, экстракт валерианы 2 драже 3 раза в день, рибоксин, тиотриазолин).
Продолжала проводиться дифференциальная диагностика с диффузными заболеваниями соединительной ткани, заболеваниями крови, вирусными и аутоиммунными поражениями печени. Попрежнему не исключалась болезнь Вильсона — Коновалова. В пользу этого диагноза свидетельствовали: молодой возраст пациента, тяжелая форма поражения печени, снижение уровня сывороточного церулоплазмина и меди крови. Однако снижение уровня церулоплазмина могло быть проявлением нарушения белковосинтетической функции печени при ее поражении и другой природы. Суточная экскреция меди с мочой оставалась нормальной. Для верификации диагноза и тактики лечения девочка была направлена в Украинскую детскую специализированную больницу, где подтвержден диагноз «болезнь Вильсона — Коновалова, тяжелая форма».
Предыдущая терапия ребенку была отменена. Назначено новое лечение: соблюдение диеты с исключением медьсодержащих продуктов (баранины, курятины, утятины, колбасы, печени, рыбы (трески), ракообразных, грибов (шампиньонов), какао, шоколада, кофе, орехов, бобовых, капусты брокколи, кресссалата, щавеля, лукапорея, редиса, чернослива, меда, перца), а также купренил (Dпеницилламин) в дозе 0,75 мг/сут, урсодезоксихолевая кислота 500 мг/сут.
Состояние ребенка значительно улучшилось, жалоб не предъявляет. Клинические проявления печеночной недостаточности отсутствуют, нормализовались показатели периферической крови и печеночного комплекса. В настоящее время наблюдается в гастроцентре, получает базисную терапию купренилом.
Особенностью данного случая является сложная диагностика наследственного заболевания обмена меди, возникшего на фоне перенесенного хронического гепатита В. Ведущими симптомами у пациентки были: поражение печени с доминированием признаков аутоиммунного процесса, выраженной печеночной недостаточности, стойкого, трудно корригируемого отечного синдрома, псевдоартритического синдрома, а также признаков энцефалопатии.
В заключение следует отметить, что болезнь Вильсона — Коновалова — редкое заболевание, которое быстро прогрессирует без адекватного лечения с развитием отечноасцитического, желтушного синдромов, печеночноклеточной недостаточности. Чаще выявляются признаки поражения печени (их характер варьирует от картины фульминантного гепатита с печеночной недостаточностью до хронического гепатита и цирроза без нарушения функции в течение длительного времени), а также поражение почек и нервнопсихические расстройства. Диагностически значимым является снижение уровня церулоплазмина в крови. У детей на ранней стадии заболевания кольцо Кайзера — Флейшера может отсутствовать. Случаи умеренных субнормальных показателей меди мочи при подозрении на БВК требуют проведения базального 24часового исследования содержания меди в моче. Важным является тот факт, что эффективное лечение купренилом позволяет предотвратить тяжелые поражения печени и других органов. В связи с этим врачу необходимо приложить максимум усилий для своевременной диагностики и назначения адекватной терапии.
1. Белоусова Е.Д. Гепатолентикулярная дегенерация (болезнь Вильсона — Коновалова) // Рос. вестн. перинатол. и педиатр. — 2009. — № 54(3). — С. 3437.
2. Ващенко В.И., Ващенко Т.Н. Церулоплазмин: от метаболита до лекарственного средства // Психофармакол. биол. наркол. — 2006 — № 6(3). — С. 12541269.
3. ИвановаСмоленская И.А., Маркова Е.Д., Илларишкин С.Н., Никольская Н.Н. Моногенные наследственные болезни центральной системы // Наследственные болезни нервной системы у детей: Руководство для врачей / Под ред. Ю.Б. Вельтищева, П.А. Темина. — М.: Медицина,1998. — С. 945.
4. Коновалов Н.В. Гепатоцеребральная дистрофия. — М.: Медгиз, 1960. — 556 с.
5.Чебатырева В.Д., Майданник В.Г. Пропедевтична педіатрія. — К., 1999. — 578 с.
6. Чуркина И.Г., Дамулин И.В., Артемьев Д.В. Гепатоцеребральная дегенерация. Клинические перспективы гастроэнтерологии, гепатологии. — 2006. — № 4. — С. 38.
7. Brewer G.J., Dick R.D., Johnson V.D. et al. Treatment of Wilson’s disease with zinc XVI: treatment during the pediatric years // J. Lab. Clin. Med. — 2005. — 137. — 191198.
8. Brewer G.J., Hedera P., Karen J.K. et al. Treatment of Wilson’s disease with ammonium tetrathiomolybdate // Arch. Neurol. — 2006. — 60. — 3.
9. Gollan J.L., Gollan J.G. Wilson disease in 2008: genetic, diagnostic and therapeutic aspects // J. Hepatol. — 2008. — 28. — 2836.
10. Hlubocka Z., Maracek Z., Linhart A. et al. Cardiac involvement in Wilson disease // J. Inherit. Metab. Dis. — 2004. — 25. — 4. — 26977.
11. Roberts E.A., Schilsky M.L. A practice guideline on Wilson disease // Hepatology. — 2006. — 147592.