Статья опубликована на с. 57-65
На рубеже XX–XXI столетий возникло понятие травматической эпидемии, что обусловлено продолжающимся ростом автомобилизации, механизации, увеличением риска техногенных катастроф, неблагоприятной криминальной ситуацией, вредными привычками, социально-психологической напряженностью в обществе. Повреждения черепа и головного мозга (ГМ) составляют более 30 % от числа всех травм, и ежегодно их число возрастает, по данным ВОЗ, не менее чем на 2 %.
В структуре причин смерти населения в Украине травматизм следует за сердечно-сосудистыми и онкологическими заболеваниями, а среди трудоспособного населения является ведущей причиной смерти и основной причиной инвалидизации, нанося обществу большой экономический и медико-социальный ущерб.
Количество черепно-мозговой травмы составляет 2–7 случаев на каждые 1000 человек ежегодно. 85–90 % составляют больные с легкой ЧМТ, 5–10 % — cо среднетяжелой и 5 % — с тяжелой травмой. Летальность среди больных варьирует от 1,3 до 4,9 %, достигая 15–30 % у больных с тяжелой травмой. В среднем ежегодно от ЧМТ в Украине погибает 2,4 чел. на 10 тыс. населения (в США — 1,8). Таким образом, ежегодно вследствие черепно-мозговой травмы умирает 11–12 тыс. человек. Пять процентов выживших страдают от хронической инвалидности, характеризующейся двигательными нарушениями, низким эмоциональным тонусом, заторможенностью, плохой памятью, нарушением концентрации внимания, эмоциональной лабильностью, повышенной возбудимостью, раздражительностью, агрессивностью и депрессией [1, 2].
При ЧМТ, с позиций патофизиологии, можно выделить несколько фаз развития повреждения мозга. Негативные исходы тяжелой черепно-мозговой травмы в основном связаны с развитием неконтролируемого вторичного повреждения тканей и нейровоспаления. Ранняя фаза повреждения, как правило, происходит в первые 24 ч после травмы и напрямую связана с повреждением тканей и расстройством физиологических функций; промежуточная фаза происходит в первые дни после ЧМТ и влечет за собой развитие нейровоспаления; поздняя фаза ассоциируется в первую очередь с когнитивными нарушениями, судорожным синдромом и эпилептогенезом и возникает в течение нескольких дней — недель после ЧМТ. В каждой из фаз существуют соответствующие методы лечения и вмешательства, нацеленные непосредственно на патофизиологические механизмы каждой фазы [3].
Первая фаза повреждения вещества ГМ характеризуется различными физическими повреждениями мозга в зависимости от локализации и механизма ЧМТ. Однако последующее повреждение мозга в первую очередь связано с развитием ишемического каскада. Нарушение энергетических процессов приводит к снижению утилизации глюкозы, накоплению лактата, уменьшению количества АТФ и снижению активности АТФ-зависимых ионных насосов, Са2+-индуцированной деполяризации, эксайтотоксичности и клеточной смерти. Ишемический каскад начинается с нарушения церебрального кровотока и оксигенации мозговой ткани [4]. В эксперименте показано, что эти процессы развиваются более интенсивно в мозге возрастных особей в сравнении с молодыми [5].
В связи с такой высокой значимостью контроля перфузии мозга для оценки развития его ишемических повреждений Brain Trauma Foundation рекомендует использование мониторинга внутричерепного давления (ВЧД) и церебрального перфузионного давления (ЦПД) у всех пациентов с тяжелой ЧМТ [6]. Тем не менее существуют доказательства, что контроль ВЧД и ЦПД не заменяет оценки истинной оксигенации ткани головного мозга [7].
Снижение кровотока и обмена кислорода в мозге способствует перестройке метаболизма с аэробного на анаэробный путь. Лактат является маркером анаэробного дыхания и накапливается в ткани, лишенной кислорода. При тяжелой черепно-мозговой травме отмечают нарушение способности глюкозы проникать в клетки мозга, а дефицит глюкозы коррелирует с ухудшением отдаленных результатов [8, 11]. Нарушение регуляции метаболизма происходит в мозге, даже если хорошо контролируются другие жизненно важные параметры [9]. Так, среди 76 успешно реанимированных пациентов с тяжелой ЧМТ и успешным контролем ВЧД у 76 % отмечали снижение уровня гликемии и у 93 % — повышенное соотношение лактат/пируват [10].
Дерегулирование церебрального метаболизма приводит к дефициту производства энергии в головном мозге. Впоследствии снижение АТФ приводит к недостаточности АТФ-зависимых ионных каналов и белков [12]. Ишемия, снижение церебрального кровотока и нарушение обмена веществ в итоге могут привести к эксайтотоксичности и клеточной смерти, включая апоптоз и некроз [13].
При исследовании методом микродиализа у пациентов с черепно-мозговой травмой определяют повышение возбуждающих аминокислот в веществе мозга, что коррелирует с вторичными повреждениями мозга. Повышение уровня глутамата связано с нарушением его поглощения посредством Са2+-зависимых каналов. Повышенная активность глутамата приводит к повышенной возбудимости и гибели нейронов. Избыток глутамата связывается с NMDA-рецепторами и способствует притоку ионов Ca2+ и Na+, активации ряда ферментов, ответственных за последующее повреждение клеток [14].
Различия в метаболизме нейромедиаторов зависят от анатомической локализации поврежденных участков мозга. Исследования пациентов с ЧМТ показали, что лобные области мозга наиболее уязвимы во время травмы. Именно здесь, в лобных областях, значительно распространены дофаминергические нейроны, что позволяет предполагать, что агонисты дофаминергических рецепторов могут улучшить результаты восстановления при нейротравме. Моноаминергические агенты (такие как амантадин) могут иметь выраженный эффект в улучшении результатов после ЧМТ. Травма ГМ может привести к изменениям в более глубинных регионах мозга в результате повреждений по механизму ускорения — замедления и с вовлечением в патологический процесс осевых структур головного мозга, а также моноаминергической системы ствола мозга. Вскоре после повреждения головного мозга существенно снижается содержание в плазме гомованилиновой кислоты — продукта деградации адренергических медиаторов. Ее низкие уровни коррелируют с тяжестью нарушения сознания. Лабораторные данные свидетельствуют, что введение моноаминергических агентов корректирует этот недостаток. В эксперименте назначение амантадина — антагониста NMDA-рецепторов способствовало выживанию пирамидальных нейронов гиппокампа [14].
Эти данные согласуются с другими исследованиями, где использование МК-801, антагониста рецепторов NMDA, снижало экспрессию нейрональной каспазы-3, NO-синтазы и дегенерацию митохондрий [15]. Последние данные свидетельствуют, что метаболиты NO могут быть надежными маркерами для тяжелой черепно-мозговой травмы [16]. NO является постоянной составляющей нейровоспалительного каскада. Внутриклеточное накопление Са2+ нарушает окислительное фосфорилирование в митохондриях, способствуя метаболическому дерегулированию при ЧМТ. Экспериментальные данные показали, что митохондриальная патология предшествует гибели нейронов [17].
В целом непосредственные физические и структурные изменения при черепно-мозговой травме негативно влияют на кровоток и оксигенацию в головном мозге. Механические повреждения и ишемия способствуют эксайтотоксическому каскаду и дерегулированию мозгового метаболизма, вызывая самые ранние патологические признаки ЧМТ. В описанной первой фазе интенсивная терапия у пациентов с ЧМТ, как правило, основывается на мерах поддержки и профилактики, в том числе контроле артериального давления и оксигенации, профилактике инфекции и тромбоза глубоких вен, обезболивании и контроле ВЧД и ЦПД. Обязательными первоочередными лечебными мероприятиями при повреждении ГМ являются [1, 18]:
1) устранение гипоксии (РаО2 > 70 мм рт.ст., SрО2 > 94 %);
2) устранение артериальной гипотензии –(АДсист > 90 мм рт.ст., а у больных со стойкой артериальной гипертензией на уровне 110–120 мм рт.ст.);
3) поддержание адекватного церебрального перфузионного давления (выше 50–70 мм рт.ст.);
4) мониторинг и коррекция внутричерепной гипертензии (не выше 20–25 мм рт.ст.);
5) контроль насыщения гемоглобина кислородом в яремной вене (> 50 %) или напряжения кислорода в тканях головного мозга (> 15 мм рт.ст.). Сатурация кислорода в луковице яремной вены (SjO2) в норме составляет 54–75 %. Нормальные значения напряжения кислорода в ткани мозга (ptiO2) составляют 25–30 мм рт.ст., а ишемический порог — 8–15 мм рт.ст.;
6) профилактика и лечение посттравматического вазоспазма.
Одновременно с решением перечисленных стратегических задач по предотвращению прогрессирования повреждений мозга должен рассматриваться вопрос нейропротективных и нейрореабилитационных мероприятий [19].
Профилактическая гипотермия является одним из вариантов патогенетической терапии острой фазы черепно-мозговой травмы. Охлаждение снижает уровень метаболизма головного мозга и замедляет ущерб, причиненный ЧМТ. При снижении температуры на каждый градус Цельсия потребление кислорода мозгом уменьшается на 5–7 %. Это способствует снижению энергозатрат при сохранении уровня оксигенации крови, приводя в соответствие мозговой метаболизм с уменьшенным уровнем мозгового кровотока. Гипотермия уменьшает врожденный иммунный ответ в экспериментальных моделях, а также демонстрирует угнетение нейровоспалительной фазы [19].
Согласно рекомендациям руководства Brain Trauma Foundation, доказательством III уровня показано, что гипотермия не приводит к снижению смертности по сравнению с нормотермией. Одновременно Brain Trauma Foundation сообщает предварительные данные, которые предполагают снижение смертности при поддержании целевой температуры в течение 48 ч, а у пациентов, получавших профилактическую гипотермию, отмечено улучшение по шкале исходов Глазго (GOS) в сравнении с нормотермическими пациентами [20]. Профилактическая гипотермия показывает спорные результаты из-за зависимости от нескольких переменных, включенных в ее успешную реализацию: температуры в момент травмы, времени развития начального охлаждения, скорости охлаждения, конечной температуры и механизма охлаждения, скорости согревания. Сочетание гипотермии с другими стратегиями лечения может увеличить преимущества. В результате отмечается потенциал для широкого применения лечебной гипотермии, но вопрос требует дополнительных исследований.
Гипербарическая оксигенотерапия (ГБО) является еще одним методом, который может использоваться в начальном периоде лечения ЧМТ. Путем увеличения парциального давления кислорода в крови происходит увеличение насыщения кислородом мозга, что, возможно, снижает повреждение тканей от вторичной ишемии и гипоксии [21]. Недавнее ретроспективное исследование показало, что применение гипербарической оксигенотерапии улучшало результаты по сравнению с контрольной группой [22]. Кроме того, проспективные исследования с использованием ГБО после стабилизации состояния пациентов также продемонстрировали эффективность методики на основании шкалы комы Глазго и шкалы исходов Глазго [23].
Мозг рассматривают в качестве «иммунно привилегированного» органа из-за наличия селективного гематоэнцефалического барьера, который препятствует проникновению многих чужеродных патогенов и иммунных медиаторов. Черепно-мозговая травма нарушает этот барьер и позволяет попадание химических передатчиков и иммунных клеток в паренхиму головного мозга. Кроме того, ЧМТ приводит к высвобождению цитокинов внутри самого мозга. Существуют многочисленные триггеры посттравматического воспаления, в том числе продукты периферической крови, ткани и клеточные обломки, простагландины, и активные формы кислорода и азота [24].
Основным измеряемым патологическим последствием нейровоспаления является повышение ВЧД. Воспаление производит два разнонаправленных воздействия на ткань головного мозга, с одной стороны, вызывая повреждение, и с другой — способствуя регенерации. После первоначальной травмы повышается ауторегуляция центральной нервной системы, приводящая к синтезу хемокинов в ответ на местное воспаление [25]. Секреция хемокинов поощряет экспрессию молекул адгезии на кровеносные сосуды, что позволяет миграцию лейкоцитов с периферии в паренхиму головного мозга. Лейкоциты и лимфоциты, попадая в центральную нервную систему, поддерживают прогрессирование воспаления. После черепно-мозговой травмы наблюдаются повышенная нейтрофильная инфильтрация, астроцитоз, отек. ЧМТ вызывает секрецию фактора некроза опухоли (TNF) астроцитами и микроглией, приводя к хроническому нейровоспалению, ухудшению пространственного обучения и нарушению памяти. TNF играет большую роль в познавательной дисфункции. Отечные изменения во время фазы воспаления при повреждении вещества ГМ тесно связаны с регулированием потока воды и ионов между внеклеточной жидкостью и глией. Понимание воспалительного каскада и его изменчивости в рамках различных форм ЧМТ имеет первостепенное значение для эффективного лечения травмы.
На сегодняшний день нет полноценных руководящих принципов для лечения нейровоспалительной фазы черепно-мозговой травмы, однако используют технологии мониторинга и тактику вторичной профилактики осложнений, а именно повышенного ВЧД. Мониторинг внутричерепного давления рекомендуется как доказательство II уровня, когда оценка по шкале комы Глазго составляет от 3 до 8 баллов и сопровождается аномальными данными КТ [26].
Доказана абсолютная противопоказанность применения для снижения ВЧД кортикостероидов, а именно метилпреднизолона. Широко известное исследование CRASH показало повышенный риск смерти у пациентов, которым вводили метилпреднизолон после травмы головного мозга [27].
Прогестерон — эндогенный стероидный гормон является вариантом фармакотерапии, который недавно привлек внимание ученых [28]. Прогестерон действует на мембраны, содержащие рецепторы прогестерона, которые экспрессируются в нейронах. В эксперименте показана его роль в нейропротекции [29]: значительно уменьшается отек головного мозга по сравнению с контролем [30]. Травма часто приводит к сосудистым повреждениям, а прогестерон увеличивает количество циркулирующих эндотелиальных клеток-предшественников после ЧМТ, что указывает на его роль в ремоделировании сосудов [31]. Он уменьшает гибель клеток [32], снижает пролиферацию астроцитов и потерю нейронов с тенденцией к улучшению памяти в эксперименте [33]. Недавнее клиническое исследование с пятидневным введением прогестерона у пациентов с черепно-мозговой травмой и уровнем сознания по шкале комы Глазго менее или равным 8 баллам привело к заметному улучшению в течение трехмесячного наблюдения по сравнению с плацебо [34]. В метаанализе выявлено, что прогестерон снижает риск смерти и тяжелой инвалидности [35].
Нейровоспаление способствует формированию отека мозга, тем самым увеличивая ВЧД. Длительное повышение внутричерепного давления может приводить к необратимым повреждениям мозга.
Использование маннита при повышении уровня ВЧД после ЧМТ в настоящее время рекомендуют в качестве доказательства II уровня при сохранении систолического артериального давления выше 90 мм рт.ст. [36]. Влияя на внутричерепное давление, маннит позволяет выполнить диагностические и лечебные интервенционные процедуры. Эффективность маннита может быть связана с уменьшением вазоконстрикции [37].
Гипертонический раствор NaCl — гиперосмолярный агент с меньшим воздействием на кровяное давление по сравнению с маннитом. Оба препарата уменьшают ВЧД, а недавний метаанализ обнаружил тенденцию в пользу гипертонического раствора натрия из-за лучшего уменьшения внутричерепного давления [38]. Он также полезен в случаях повышенного ВЧД, которое не реагирует на другие виды терапии. Гипертонический NaCl значительно повышает снабжение мозга кислородом по сравнению с маннитом [39].
Несколько более спорной процедурой в управлении внутричерепным давлением является декомпрессия черепа. Исследования показали позитивные результаты у более чем 50 % пациентов с тяжелой ЧМТ [40]. Тем не менее процедура сохраняет высокую смертность — 26,4 %, особенно с увеличением возраста пациента и снижением оценки по шкале комы Глазго [41]. В дополнение к смертности декомпрессия связана с многочисленными неблагоприятными исходами, в том числе с расширением зоны ушиба, новой субдуральной или эпидуральной гематомой с противоположной стороны, ликвореей, эпилепсией, церебральной грыжей, субдуральным выпотом и инфекцией. Использование этой хирургической тактики требует высокого хирургического мастерства и опыта.
Судороги являются одним из наиболее заметных долгосрочных последствий черепно-мозговой травмы, прогрессируя в эпилепсию в более тяжелых случаях. Первоначальная клеточная травма, вызванная ЧМТ, способствует гибели клеток и воспалению, которые играют роль в эпилептогенезе [42]. В простом смысле судороги развиваются, когда возбуждающие потенциалы становятся активнее тормозных потенциалов. Судороги после повреждения вещества ГМ встречаются приблизительно у 50 % больных в течение 15 лет после проникающей травмы. Есть множество факторов повышенного риска посттравматических судорог, в том числе: ШКГ < 10 баллов, корковая контузия, вдавленный перелом черепа, субдуральная или эпидуральная гематома, внутримозговая гематома, проникающие раны головы и судороги в течение первых 24 ч травмы [43, 44].
Не существует рекомендаций I уровня для противосудорожной профилактики. Приемлемы рекомендации II уровня, которые предлагают использовать противосудорожные препараты, такие как фенитоин и вальпроат, чтобы предотвратить ранние припадки, но не поздние приступы из-за побочных эффектов, связанных с хроническим использованием [45]. Другие противосудорожные препараты, такие как фенобарбитал и карбамазепин, как правило, избегают назначать из-за побочных эффектов и фармакодинамического профиля [46]. Продолжаются испытания ряда новых препаратов (леветирацетам, этосуксимид). Хирургический вариант лечения посттравматической эпилепсии — имплантация стимулятора блуждающего нерва — основан на изменении уровня норадреналина и повышении уровня ГАМК [47, 48].
Наиболее серьезными последствиями тяжелой черепно-мозговой травмы являются длительно сохраняющиеся нарушения сознания, которые могут проявляться в виде вегетативного состояния и минимального уровня сознания (minimally conscious state).
Нарушения сознания при ЧМТ состоят из нарушений уровня сознания (бодрствования) и содержания сознания (когнитивные и эмоционально-психические функции). Бодрствование поддерживают несколько популяций стволовых нейронов, которые имеют проекцию на таламические и кортикальные структуры. Тяжелое повреждение ствола и/или обоих полушарий мозга может вызвать снижение уровня бодрствования. Однако нарушение стволовых рефлексов может сочетаться с интактной функцией активирующей ретикулярной формации, если покрышка пероральных отделов ствола и мезенцефалон не повреждены. Считают, что осознанность обеспечивается интегративной деятельностью коры и ее подкорковых связей [49].
При вегетативном статусе пациенты могут бодрствовать, но они не осознают самих себя и окружающий мир. Термин «вегетировать» введен основоположниками изучения нарушений сознания Б. Дженнеттом и Ф. Пламом для обозначения способности «жить, как биологический организм, исключая интеллектуальную активность и социальное общение». Как «вегетирующий» описывают «организм, способный к росту и развитию, исключая восприятие и мышление». Характерные симптомы вегетативного статуса, согласно описанию Б. Дженнетта и Ф. Плама: отсутствие клинически значимого адаптивного ответа на окружающую действительность; отсутствие признаков деятельности разума относительно восприятия или передачи информации; продолжительные периоды бодрствования; относительная акинезия (возможны вынужденные позы и примитивное стереотипное отдергивание); пациент не разговаривает, но способен издавать отдельные звуки; нет способности сигнализировать движениями глазных яблок, хотя иногда возможно медленное и неустойчивое слежение за движущимся предметом; первичная ЭЭГ может выглядеть изолинией, но спустя несколько месяцев может регистрироваться некоторая активность.
Согласно заключению международной группы экспертов (Aspen Neurobehavioral Conference Workgroup, 1995–2000), состояние минимального сознания характеризует пациентов, которые не находятся в вегетативном статусе, но длительно не способны к полному контакту. Эти больные демонстрируют ограниченные и непостоянные, но явные и воспроизводимые признаки ориентировки в самих себе или в окружающем их пространстве. Обязательно наличие хотя бы одного из следующих критериев: выполнение простых команд, жестовый или вербальный ответ «да/нет» (невзирая на правильность), осознанная речь, осмысленное поведение (включая движения или эмоциональные реакции в ответ на внешние раздражители). Необходимо убедиться в максимальном уровне бодрствования (адекватная стимуляция) и исключить афазию, агнозию, апраксию и сенсомоторный дефицит как причину недостаточной коммуникации. Дальнейшее уменьшение неврологического дефицита у таких пациентов значительно более вероятно, чем при вегетативном статусе [50].
В синдромокомплексе травматической болезни головного мозга важное место занимают расстройства когнитивной сферы. Когнитивный дефицит той или иной степени выраженности отмечается при всех типах черепно-мозговой травмы. При тяжелых ушибах лобных и височных долей, диффузном аксональном повреждении обычно развивается первичная посттравматическая деменция, которая проявляется сразу после выхода пациента из комы.
Механизмы развития когнитивных расстройств в остром периоде ЧМТ включают окислительный стресс, эксайтотоксичность, перифокальную деполяризацию нейронов, аутоиммунное воспаление и апоптоз. Все эти механизмы в конечном итоге ведут к гибели нейронов и потере ассоциативных связей между различными отделами центральной нервной системы. Задача лечения пациентов в остром периоде ЧМТ как раз и состоит в прерывании этих патологических процессов и стимуляции естественных механизмов защиты нейронов от повреждения посредством назначения корректоров гемодинамики, метаболических субстратов, антиоксидантов и других нейрореабилитационных технологий.
Проведено много исследований препаратов с потенциально нейропротективным воздействием для блокирования каскада вторичного повреждения мозговой ткани. Гипотезы, которые поддерживаются в данных исследованиях, направлены на скаведжинг свободных радикалов, антиоксидантное и противовоспалительное воздействие, ингибирование поступления свободных ионов кальция в нейроны различными механизмами. Однако практически все большие клинические испытания в этой области были малоуспешными [18, 50]. Сегодня продолжается поиск молекул и их комбинаций, способных эффективно ингибировать каскадные механизмы гибели нейронов и компонентов нейроглии, которые запускаются в зоне ишемического повреждения ткани мозга при черепно-мозговой травме.
Описан целый ряд механизмов реализации нейропротекции (церебропротекции), в том числе снижение энергетических затрат и потребности мозговой ткани в кислороде, восстановление энергетических ресурсов; антиоксидантные реакции; антагонизм по отношению к глутаматным рецепторам, ингибиция синтеза и пресинаптического высвобождения глутамата; агонистическое действие с ГАМК, глицином; антагонистическое отношение к потенциалзависимым Са-каналам; модуляция нейрональной NO-синтазы; блокада холинэстеразы и К-каналов; ангиопротекция; повышение устойчивости к гипоксии [51].
В нашей практике мы придерживаемся описанных выше рекомендаций для целенаправленной интенсивной терапии как основы лечения тяжелой ЧМТ, как изолированной, так и в комплексе политравматических повреждений. В комплексе нейропротекции и нейрореабилитации при тяжелой ЧМТ, риске развития вегетативных состояний и синдромах малого сознания с 2012 г. мы активно применяем оригинальный амантадина сульфат (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») в инфузионной и таблетированной формах выпуска.
Амантадин в виде водорастворимой соли впервые был использован в качестве противовирусного агента в 1960 году. С 1980-х годов препарат используют для лечения симптомов паркинсонизма у пациентов, перенесших черепно-мозговую травму, а также при болезни Паркинсона [52]. Амантадин действует в качестве антагониста N-метил-D-аспартата и косвенного агониста допамина. Он обладает способностью проникать во все клеточные мембраны, включая центральную нервную систему. Хотя механизм действия до сих пор не совсем понятен, амантадин, как полагают, облегчает высвобождение дофамина пресинаптическими нейронами и его связывание с постсинаптическими рецепторами [53]. Амантадин, как антагонист рецепторов N-метил-D-аспартата, может обеспечить защиту от вторичного повреждения в результате высвобождения глутамата. Этот препарат представлен в литературе в качестве варианта терапии для улучшения психического статуса при ЧМТ. Амантадин является одним из часто назначаемых препаратов для пациентов с длительными расстройствами сознания после ЧМТ в связи с тем, что может улучшать функциональное восстановление [54].
Описана высокая эффективность препарата в отношении терапии симптомов дисфункции лобных долей при ЧМТ и других видах приобретенного повреждения головного мозга (инсульт, аневризмы). Эти симптомы могут включать в себя проблемы с краткосрочной памятью, вниманием, планированием решения проблем, импульсивность, расторможенность, слабую мотивацию и другие поведенческие и когнитивные нарушения (синдром лобной доли) [55–57].
В 2012 г. в многоцентровом двойном слепом рандомизированном плацебо-контролируемом исследовании было показано, что амантадина сульфат ускоряет восстановление сознания после тяжелой ЧМТ. Исследование проведено в 11 клиниках трех стран. Критерии включения: возраст 16–65 лет, непроникающая ЧМТ, 4–16 недель после травмы, вегетативное состояние или минимальный уровень сознания (оценка по шкале DRS (Disability Rating Scale) более 11 баллов), неспособность к выполнению команд, общению (оцениваемые по шкале CRS-R (Coma Recovery Scale-Revised)).
Амантадина сульфат в таблетированной форме выпуска назначали по 100 мг дважды в сутки в течение 14 дней. Затем дозу препарата увеличивали до 150 мг дважды в сутки на протяжении третьей недели, затем — до 200 мг дважды в день на протяжении четвертой недели. В течение четырехнедельного курса терапии сознание, по данным шкалы DRS, в исследуемой группе восстанавливалось быстрее, чем в группе плацебо (Р = 0,007). Препарат был эффективен у больных в вегетативном состоянии и с минимальным уровнем сознания. Побочных эффектов препарат не вызывал.
Положительный эффект амантадина сульфата может обусловливаться увеличением передачи импульса в допаминзависимых нейрональных регионах центральной нервной системы, связанных с уровнем сознания. Влияние препарата на длительные исходы заболевания остается неясным. Амантадин обладает свойствами антагониста N-метил-D-аспартат-рецепторов и агониста рецепторов к допамину. В остром периоде после ЧМТ тяжелой степени наблюдается кратковременный период гипервозбудимости нейронов, что в последующем сменяется гипоактивностью нейронов и уменьшением нейротрансмиттеров центральной нервной системы, включая допамин. Амантадина сульфат может увеличивать допаминергическую активность нервной системы посредством ускорения пресинаптического выделения медиатора и блокады повторного постсинаптического захвата. Положительный эффект препарата может обусловливаться увеличением передачи импульса в допаминзависимых нейрональных регионах центральной нервной системы, связанных с уровнем сознания [58].
В мультицентровом проспективном нерандомизированном открытом исследовании по типу «до и после» проведено изучение эффективности оригинального амантадина сульфата (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») в инфузионной и таблетированной формах выпуска у пациентов с тяжелой церебральной недостаточностью.
В отделении интенсивной терапии политравмы КУ «Днепропетровская областная клиническая больница им. И.И. Мечникова» проведено проспективное наблюдательное исследование эффективности и безопасности оригинального амантадина сульфата (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») у пациентов в возрасте 20–65 лет с тяжелой травмой головного мозга, которая являлась превалирующей в составе политравмы. Обследовано 30 больных. Критериями отбора были оценка по шкале комы Глазго 10 баллов и меньше в течение первых 24 ч после травмы и неблагоприятная КТ-картина травмы мозга. Пациенты были разделены в зависимости от назначенной терапии на 2 группы по 15 человек, 1-я из которых (n = 15) получала терапию согласно стандартному протоколу лечения, больным 2-й группы (n = 15) в дополнение к стандартной терапии назначали амантадина сульфат (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») в дозе 200 мг внутривенно в течение 3–5 дней, затем переходили на таблетированную форму препарата, назначая его в дозе 200 мг 2 раза в сутки с 4-х по 10-е сутки, 200 мг 1 раз в сутки — с 11-х по 28-е сутки заболевания.
Эффективность терапии оценивали на основании:
— определения тяжести состояния по шкалам TS, ISS, GCS, RLAS, APACHE II;
— учета потребности в искусственной вентиляции легких и ее продолжительности;
— уровня летальности до 28-го дня.
Безопасность терапии оценивали путем мониторинга уровня электролитов и оценки наличия или отсутствия существенных побочных эффектов стимуляции центральной нервной системы. Ни у одного пациента не отмечено расстройств электролитного баланса в течение периода наблюдения, а также не было зарегистрировано признаков перевозбуждения центральной нервной системы (бессонница, нервозность и гиперактивность).
Проведена статистическая обработка полученных данных.
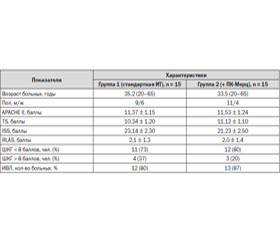
Сравнительная исходная характеристика групп больных представлена в табл. 1.
По демографическим данным, клиническим характеристикам, степени тяжести исходного состояния при оценке по общепринятым шкалам травмы и неврологического дефицита, а также по потребности в проведении искусственной вентиляции легких группы больных исходно были идентичны.
Данные об оценке эффективности лечения в исследуемых группах больных представлены в табл. 2.
Выявлено позитивное влияние оригинального амантадина сульфата (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») в инфузионной и таблетированной формах выпуска на восстановление качества жизни больных, что проявлялось уменьшением времени достижения VI уровня шкалы RLAS на 21 %, скорости восстановления уровня сознания по шкале комы Глазго > 12 баллов на 14 %.
Количество больных, нуждающихся в искусственной вентиляции легких длительностью более 7 дней, в подгруппе амантадина было меньше на 7 %, что могло быть связано с более быстрым улучшением неврологического статуса.
В исследуемой группе отмечено снижение летальности до 28-го дня наблюдения на 4 % по сравнению с контрольной группой.
Выводы
Интенсивная терапия тяжелой черепно-мозговой травмы является ведущей в комплексе лечебных мероприятий у пациентов с сочетанной травмой. Она основывается на патогенетическом подходе и достижении требуемых (целевых) параметров перфузии и оксигенации мозга с обязательным контролем ВЧД. Применение блокаторов глутаматных NMDA-рецепторов и влияние на глутаматную активность являются обоснованными.
Клинический опыт применения оригинального амантадина сульфата (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») у пациентов с тяжелой ЧМТ показал:
— снижение летальности в группе, получавшей ПК-Мерц®, «Мерц Фарма ГмбХ и Ко», по сравнению с контрольной группой пациентов на 4 %;
— уменьшение длительности пребывания пациентов, получавших ПК-Мерц®, «Мерц Фарма ГмбХ и Ко», на искусственной вентиляции легких на 7 %;
— более быструю динамику восстановления неврологического статуса пациентов, получавших ПК-Мерц®, «Мерц Фарма ГмбХ и Ко», что клинически проявлялось уменьшением скорости восстановления уровня сознания по шкале комы Глазго на 14 % и тяжести состояния по шкале RLAS на 21 % в пользу группы с ПК-Мерц®, «Мерц Фарма ГмбХ и Ко»;
— применение оригинального амантадина сульфата (ПК-Мерц®, «Мерц Фарма ГмбХ и Ко») в инфузионной и таблетированной формах выпуска не оказало влияния на электролитный баланс плазмы крови, а также не сопровождалось признаками возбуждения;
— возможность снижения глутаматной активности и уменьшения повреждающих факторов эксайтотоксичности при ЧМТ позволяет влиять на прогноз выживаемости пациента.
ПК-Мерц®, «Мерц Фарма ГмбХ и Ко», рекомендовано вводить в первые 12 часов от начала заболевания в/в 1 раз в сутки со скоростью 30 капель в 1 минуту в дозе 200 мг (1 флакон по 500 мг) в течение 3–5 дней заболевания, затем переходить на таблетированную форму препарата, назначая его в дозе 200 мг 2 раза в сутки с 4-х по 10-е сутки, 200 мг 1 раз в сутки — с 11-х по 28-е сутки заболевания.
Список литературы
1. Педаченко Є.Г. Сучасні принципи та стан надання невідкладної допомоги при черепно-мозковій травмі в Україні / Матеріали конференції нейрохірургів України «Актуальні питання невідкладної нейрохірургії» 21–23 вересня 2005 року, м. Тернопіль // Український нейрохірургічний журнал. — 2005. — № 3. — С. 4-6.
2. Овсянников Д.М., Чехонацкий А.А., Колесов В.Н., Бубашвили А.И. Социальные и эпидемиологические аспекты черепно-мозговой травмы (обзор) // Саратовский научно-медицинский журнал. — 2012. — Т. 8, № 3. — С. 777-785.
3. Algattas Н., Huang J.H. Traumatic Brain Injury Pathophysio–logy and Treatments: Early, Intermediate, and Late Phases Post-Injury // Int. J. Mol. Sci. — 2014. — 15(1). — 309-341.
4. Leung L.Y., Wei G., Shear D.A., Tortella F.C. The acute effects of hemorrhagic shock on cerebral blood flow, brain tissue oxygen tension, and spreading depolarization following penetrating ballistic-like brain injury // J. Neurotrauma. — 2013. — 30. — 1288-1298.
5. Hawkins B.E., Cowart J.C., Parsley M.A. et al. Effects of trauma, hemorrhage and resuscitation in aged rats // Brain Res. — 2013. — 1496. — 28-35.
6. Bratton S.L., Chestnut R.M., Ghajar J. et al. Guidelines for the management of severe traumatic brain injury. IX. Cerebral perfusion thresholds // J. Neurotrauma. — 2007. — 24. — S59-S64.
7. Eriksson E.A., Barletta J.F., Figueroa B.E. et al. Cerebral perfusion pressure and intracranial pressure are not surrogates for brain tissue oxygenation in traumatic brain injury // Clin. Neurophysiol. — 2012. — 123. — 1255-1260.
8. Glenn T.C., Kelly D.F., Boscardin W.J. et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism // J. Cereb. Blood Flow Metab. — 2003. — 23. — 1239-1250.
9. Selwyn R., Hockenbury N., Jaiswal S. et al. Mild traumatic brain injury results in depressed cerebral glucose uptake: An FDG PET study // J. Neurotrauma. — 2013. — 30. — 1943-1953.
10. Stein N.R., McArthur D.L., Etchepare M., Vespa P.M. Early cerebral metabolic crisis after TBI influences outcome despite ade–quate hemodynamic resuscitation // Neurocrit. Care. — 2012. — 17. — 49-57.
11. Matsushima K., Peng M., Velasco C. et al. Glucose variability negatively impacts long-term functional outcome in patients with traumatic brain injury // J. Crit. Care. — 2012. — 27. — 125-131.
12. Werner C., Engelhard K. Pathophysiology of traumatic brain injury // Br. J. Anaesth. — 2007. — 99. — 4-9.
13. Raghupathi R., Graham D.I., McIntosh T.K. Apoptosis after traumatic brain injury // J. Neurotrauma. — 2000. — 17. — 927-938.
14. Wang T., Huang X.J., Van K.C. аt al. Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats // J. Neurotrauma. — 2013. — doi:10.1089/neu.2013.2917.
15. Han R.Z., Hu J.J., Weng Y.C. et al. NMDA receptor antagonist MK-801 reduces neuronal damage and preserves learning and memory in a rat model of traumatic brain injury // Neurosci. Bull. — 2009. — 25. — 367-375.
16. Kandasamy R., Kanti Pal H., Swamy M., Abdullah J. Cerebrospinal fluid nitric oxide metabolite levels as a biomarker in severe traumatic brain injury // Int. J. Neurosci. — 2013. — 123. — 385-391.
17. Singh I.N., Sullivan P.G., Deng Y. et al. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy // J. Cereb. Blood Flow Metab. — 2006. — 26. — 1407-1418.
18. Черний Т.В., Стецик В.Ю., Черний В.И. Черепно-мозговая травма в аспекте доказательной медицины: обзор актуальных международных рекомендаций // Медицина неотложных состояний. — 2014. — 5(60).
19. Tomura S., de Rivero Vaccari J.P., Keane R.W. et al. Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury // J. Cereb. Blood Flow Metab. — 2012. — 32. — 1939-1947.
20. Bratton S.L., Chestnut R.M., Ghajar J. et al. Guidelines for the management of severe traumatic brain injury. III. Prophylactic hypothermia // J. Neurotrauma. — 2007. — 24. — S21-S25.
21. Huang L., Obenaus A. Hyperbaric oxygen therapy for traumatic brain injury // Med. Gas Res. — 2011. — 1. — 21.
22. Sahni T., Jain M., Prasad R. et al. Use of hyperbaric oxygen in traumatic brain injury: Retrospective analysis of data of 20 patients treated at a tertiary care centre // Br. J. Neurosurg. — 2012. — 26. — 202–207.
23. Lin J.W., Tsai J.T., Lee L.M. et al. Effect of hyperbaric oxygen on patients with traumatic brain injury // Acta Neurochir. –Suppl. — 2008. — 101. — 145-149.
24. Dardiotis E., Karanikas V., Paterakis K. et al. Traumatic Brain Injury and Inflammation: Emerging Role of Innate and Adaptive Immunity // Brain Injury — Pathogenesis, Monitoring, Recovery and Management; Agrawal A., Ed. — InTech: Rijeka, Croatia, 2012.
25. Ransohoff R.M. The chemokine system in neuroinflammation: An update // J. Infect. Dis. — 2002. — 186. — S152-S156.
26. Bratton S.L., Chestnut R.M., Ghajar J. et al. Guidelines for the management of severe traumatic brain injury. VII. Intracranial pressure monitoring technology // J. Neurotrauma. — 2007. — 24. — S45-S54.
27. Roberts I., Yates D., Sandercock P. et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial // Lancet. — 2004. — 364. — 1321-1328.
28. Attella M.J., Nattinville A., Stein D.G. Hormonal state affects recovery from frontal cortex lesions in adult female rats // Behav. Neural boil. — 1987. — 48. — 352-367.
29. Meffre D., Labombarda F., Delespierre B. et al. Distribution of membrane progesterone receptor alpha in the male mouse and rat brain and its regulation after traumatic brain injury // Neuroscience. — 2013. — 231. — 111-112.
30. Wright D.W., Bauer M.E., Hoffman S.W., Stein D.G. Serum progesterone levels correlate with decreased cerebral edema after traumatic brain injury in male rats // J. Neurotrauma. — 2001. — 18. — 901-909.
31. Li Z., Wang B., Kan Z. et al. Progesterone increases circula–ting endothelial progenitor cells and induces neural regeneration after traumatic brain injury in aged rats // J. Neurotrauma. — 2012. — 29. — 343-353.
32. Barha C.K., Ishrat T., Epp J.R. et al. Progesterone treatment normalizes the levels of cell proliferation and cell death in the dentate gyrus of the hippocampus after traumatic brain injury // Exp. Neurol. — 2011. — 231. — 72-81.
33. Tang H., Hua F., Wang J. et al. Progesterone and vitamin D: Improvement after traumatic brain injury in middle-aged rats // Horm. Behav. — 2013. — 64. — 527-538.
34. Shakeri M., Boustani M.R., Pak N. et al. Effect of progesterone administration on prognosis of patients with diffuse axonal injury due to severe head trauma // Clin. Neurol. Neurosurg. — 2013. — 115. — 2019-2022.
35. Ma J., Huang S., Qin S., You C. Progesterone for acute traumatic brain injury // Cochrane Database Syst. Rev. — 2012. — 10. — doi:10.1002/14651858.CD008409.pub3.
36. Bratton S.L., Chestnut R.M., Ghajar J. et al. Guidelines for the management of severe traumatic brain injury. II. Hyperosmolar therapy // J. Neurotrauma. — 2007. — 24. — S14-S20.
37. Muizelaar J.P., Lutz H.A., 3rd, Becker D.P. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients // J. Neurosurg. — 1984. — 61. — 700-706.
38. Rickard A.C., Smith J.E., Newell P. et al. Salt or sugar for your injured brain? A meta-analysis of randomised controlled trials of mannitol versus hypertonic sodium solutions to manage raised intracranial pressure in traumatic brain injury // Emerg. Med. J. — 2013. — doi:10.1136/emermed-2013-202679.
39. Oddo M., Levine J.M., Frangos S. et al. Effect of mannitol and hypertonic saline on cerebral oxygenation in patients with severe traumatic brain injury and refractory intracranial hypertension // J. Neurol. Neurosurg. Psychiatry — 2009. — 80. — 916-920.
40. Williams R.F., Magnotti L.J., Croce M.A. et al. Impact of decompressive craniectomy on functional outcome after severe traumatic brain injury // J. Trauma. — 2009. — 66. — 1570-1574.
41. Huang Y.H., Lee T.C., Lee T.H. et al. Thirty-day mortality in traumatically brain-injured patients undergoing decompressive craniectomy // J. Neurosurg. — 2013. — 118. — 1329-1335.
42. Tomkins O., Feintuch A., Benifla M. et al. Blood-brain barrier breakdown following traumatic brain injury: A possible role in posttraumatic epilepsy // Cardiovasc. Psychiatry Neurol. — 2011.
43. Temkin N.R., Dikmen S.S., Wilensky A.J. et al. A rando–mized, double-blind study of phenytoin for the prevention of post-traumatic seizures // N. Engl. J. Med. — 1990. — 323. — 497-502.
44. Yablon S.A. Posttraumatic seizures // Arch. Phys. Med. Rehabil. — 1993. — 74. — 983-1001.
45. Bratton S.L., Chestnut R.M., Ghajar J. et al. Guidelines for the management of severe traumatic brain injury. XIII. Antiseizure prophylaxis // J. Neurotrauma. — 2007. — 24. — S83-S86.
46. Torbic H., Forni A.A., Anger K.E. et al. Use of antiepileptics for seizure prophylaxis after traumatic brain injury // Am. J. Health-Syst. Pharm. — 2013. — 70. — 759-766.
47. Ghanem T., Early S.V. Vagal nerve stimulator implantation: An otolaryngologist’s perspective // Otolaryngol. Head Neck Surg. — 2006. — 135. — 46-51.
48. Algattas H., Huang J.H. Neurotrauma and repair research: Traumatic brain injury (TBI) and its treatments // Biomed. Eng. Comput. Biol. — 2013. — 5. — 51-56.
49. Яворская В.А., Лысенко В.И., Фломин Ю.В. и др. Диагностика и лечение вегетативного состояния в клинической практике // Острые и неотложные состояния в практике врача. — 2006. — № 1(01). — С. 7-14.
50. Челяпина М.В., Шарова Е.В., Зайцев О.С. Cиндром дофаминергической недостаточности в картине тяжелой травмы мозга на фоне длительного угнетения сознания // Неврология, нейропсихиатрия, психосоматика. — 2014. — № 4.
51. Евтушенко И.С. ноотропы и нейропротекторы в современной клинической нейрофармакологии // Международный неврологический журнал. — 2013. — 3(57).
52. Кraus M., Maki P. Case report: the combined use of amantadine and l-dopa/carbidopa in the treatment of chronic brain injury // Brain Injury. — 1997. — 11. — 455-460.
53. Кarl D., Burke D., Kim H. et al. Effects of dopaminergic combination in traumatic brain injury rehabilitation // Brain Injury. — 1999. — 13. — 63-68.
54. Leone H., Polsonetti B.W. Amantadine for traumatic brain injury: does it improve cognition and reduce agitation? // J. Clin. Pharm. Ther. — 2005. — 30(2). — 101-104.
55. Kraus M.F., Maki P.M. Effect of amantadine hydrochloride on symptoms of frontal lobe dysfunction in brain injury: case studies and review // J. Neuropsychiatry Clin. Neurosci. — 1997. — 9(2). — 222-230.
56. Meythaler J.M., Brunner R.C., Johnson A., Novack T.A. Amantadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury: a pilot double-blind randomized trial // J. Head Trauma Rehabil. — 2002. — 17(4). — 300-313.
57. Voit M. Does Treatment with Amantadine Increase the Rate of Improvement of Cognitive Function in Patients Suffering from Traumatic Brain Injury? — 2014 // PCOM Physician Assistant Studies Student Scholarship. — Paper 198.
58. Giacino J.T., Whyte J., Bagiella E. et al. Placebo-controlled trial of amantadine for severe traumatic brain injury // N. Engl. J. Med. — 2012. — V. 366. — P. 819-826.

/63.jpg)