Статья опубликована на с. 24-29
В настоящее время в клинической практике при критических состояниях, сопровождающихся гипоксией головного мозга различного генеза, в комплексе интенсивного лечения используется фармакологическая нейропротекция. При этом эффекты NMDA-рецепторов рассматривают только как источник триггерной активности эксайтотоксичности, без учета локализации их структуры, физиологических аспектов и патофизиологических последствий в плане влияния на состояние когнитивных функций.
Вместе с тем появление новой информации об особенностях функционирования глутаматергической системы требует более углубленного понимания различий механизмов реализации ее отдельных интегративных элементов. Это имеет важное значение не только для более полного представления патогенетических процессов, протекающих в ишемизированных отделах головного мозга, но и для разработки дифференцированного фармакологического обеспечения нейрональной жизнедеятельности, расширения диапазона нейропротективных возможностей самой целлюлярной системы. К тому же это касается и фармакологического контроля нейропластичности и нейрорегенерации, при котором именно глутаматергическая система играет существенную роль. С этой точки зрения рассмотрение появившейся информации о функциональной активности NMDA-рецепторов в зависимости от их экстрасинаптической или синаптической локализации может помочь в реализации дифференцированного подхода к разработке метаботропной терапии острого периода состояний, связанных с ишемией/реперфузией ткани головного мозга, с учетом сохранения когнитивных функций.
С позиции вышеприведенных представлений следует напомнить о некоторых существенных отличительных моментах селективности сигналинговых путей, опираясь на исследования, раскрывающие сущность этих различий в зависимости от модулирующего влияния (активация или угнетение) на баланс взаимоотношений между экстрасинаптическим и синаптическим пулами NMDA-рецепторов, которые и определяют два противоположных эффекта — активацию процессов гибели клеток или поддержание их жизнедеятельности.
При этом заслуживают внимания несколько следующих основных биохимических направлений:
— особенности действия подтипов экстрасинаптических и синаптических NMDA-рецепторов;
— инфлюкс ионов Са++ и направленность его потока во взаимосвязи с системой модулирующих компонентов ядра клетки или без них;
— включение и активирование соответствующих трансдукционных и транскрипционных факторов вследствие влияния на митогенактивируемые протеинкиназы (МАРК), преимущественно через экстрацеллюлярную сигналинговую протеинкиназу (ERK или Srk — Signaling regulated kinase) [1–3].
Исходя из различий пула NMDA-структур по локализации, целесообразно иметь представление об особенностях их функциональной дифференцированности.
Ультраструктурные особенности экстрасинаптических NMDA-рецепторов и соответствующие биохимические процессы
Экстрасинаптические NMDA-рецепторы в отличие от синаптических располагаются на нейронах и дендритах, на концах шипиков дендритных отростков, концентрируемых в местах контактов с соседними или смежными отростками, которые являются преимущественно аксонами, терминалями аксонов или клетками глии [4, 5]. В местах ассоциаций с соседними отростками имеются характерные постсинаптические уплотнения.
Говоря о функциональной активности экстрасинаптических NMDA-рецепторов, следует отметить, что они активируются высокочастотными импульсами с частыми квантами глутамата в отличие от синаптических структур NMDA, где адекватным является низкочастотный характер импульсов, инициируемый единичными квантами глутамата [5].
Установлено, что дендритный сигнал при участии потенциалзависимых каналов, локализованный как на самом шипике дендрита в непосредственной близости от синапса, как и в субсинаптической зоне на дендритных отростках, играет важную роль в модуляции локальных физиологических функций не только дендрита, но и нейрона в целом [6].
Однако дальнейшая активация экстрасинаптических NMDA-рецепторов в условиях прогрессирования ишемии/реперфузии ткани головного мозга становится ключевым фактором в развитии эксайтотоксического повреждения [7]. Это проявляется в первую очередь деструктивными изменениями со стороны дендритов, набуханием нейронов и выраженным скоротечным снижением потенциала митохондриальных мембран, что сопровождается дисфункцией митохондрий и активацией программы клеточной гибели.
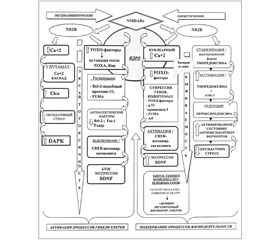
В основе этого процесса лежат тонкие биохимические механизмы при участии ряда факторов, связанных с программой гибели клеток и непосредственно обусловленных активностью именно экстрасинаптических NMDA-рецепторов, реализуемой через их подтип NR2B. Последний обладает свойством универсальности, так как поддержание процессов жизнедеятельности в клетке связано с этим же подтипом, но с локализацией его в синаптических структурах NMDA-рецепторов (рис. 1).
Среди ряда транскрипционных факторов наиболее значимым является нуклеарный комплекс FOXO (forkhead box O), который представляет собой комплекс транскрипционных факторов, проявляющий свою активность в условиях эксайтотоксичности и оксидативного стресса [5]. Его действие направлено на инициирование соответствующих целевых генов, кодирующих продукцию факторов, которые обусловливают блокирование активации интрацеллюлярной антиоксидантной системы и препятствуют ингибированию апоптоза. Этот механизм ассоциирован с активацией экстрасинаптического пула NMDA-рецепторов и опосредуется транслокацией FOXO из цитоплазмы в ядро, что рассматривается наравне со стимулирующими сигналами, инициирующими гибель клетки, к которым относятся прекращение влияния ростовых факторов и супрессия аэробного метаболизма [8] (рис. 1).
Кроме того, в условиях церебральной ишемии при сохранении активности подтипа NR2B в экстрасинаптических NMDA-рецепторах активизируется специфическая протеинкиназа, действие которой также ассоциировано с процессом нейрональной гибели, — DAPK [9] (рис. 1). При этом усиливается процесс фосфорилирования самой структуры NR2B экстрасинаптических NMDA-рецепторов, благодаря чему развертываются процессы усиленного инфлюкса Са++ по каналам, ассоциированным с экстрасинаптическими NMDA-рецепторами.
Еще одним из механизмов реализации повреждающего эффекта глутаматассоциируемой эксайтотоксичности при стимуляции экстрасинаптических NMDA-рецепторов является включение кальцийактивируемых хлоридных каналов Сlca-1 [10], которые в комплексе с FOXO рассматриваются как часть реализации геномной программы нейрональной гибели, запускаемой через активацию экстрасинаптических NMDA-рецепторов [5, 10, 11] (рис. 1).
Немаловажным является и то, что при активации экстрасинаптического пула NMDA-рецепторов имеет место выключение сигналингового пути функционирования соединения СREB (cAMP response element binding protein — белок, связывающий цАМФ-отвечающие элементы ДНК через цАМФ-сигнальный каскад) [12]. Это соединение является посредником нейрорегенерации и неотъемлемым компонентом в реализации эффекта нейротрофинов и ростовых факторов, включая BDNF.
Однако в условиях гипоксии/ишемии с активацией экстрасинаптических NMDA-рецепторов выключение СREB блокирует экспрессию ростового фактора BDNF (рис. 1) [13], что приводит к крайнему замедлению процессов нейропластичности и нейрорегенерации. При этом сохраняется активный инфлюкс Са++, снижение мембранного потенциала митохондрий с последующей активацией процессов клеточной смерти.
Таким образом, при острой ишемии/реперфузии тканей головного мозга повышается активность NMDA-рецепторов экстрасинаптической локализации, инициирующих комплекс геномного пула апоптотических ядерных факторов. Это, в свою очередь, ведет к блокированию активности антиапоптотических факторов, экспрессии нейротрофинов, что в условиях прогрессирующего анаэробного метаболизма запускает программу гибели клеток.
Синаптическая активность NMDA-рецепторов
Иную направленность имеет активация синаптического пула NMDA-рецепторов — это поддержание жизнедеятельности и нейропротективного эффекта, что является достаточно значимой биологической целесообразностью его рецепторного сигналинга.
Отличие синаптического NMDA-эффекта от экстрасинаптического обусловлено рядом следующих факторов, способствующих активации синаптических NMDA-рецепторов:
— супрессия процесса продуцирования комплекса факторов FOXO и выведение из ядра нейрона (нуклеарный экспорт) [14] (рис. 1);
— активация фосфорилирования компонентов FOXO, в связи с чем последние утрачивают активность в отношении поддержания процессов гибели клетки;
— пролонгация ингибирования целевых генов, кодирующих появление факторов FOXO;
— контроль устойчивости и стабильности функционирования системы глутатиона и нейрональной уязвимости к разрушительному влиянию оксидативного стресса [5].
Важно подчеркнуть, что в сохранении активированного состояния антиоксидантных ферментов большое значение имеет стабилизация восстановленной формы тиоредоксина, которая усиливается посредством возбуждения синаптических NMDA-рецепторов, и этим же путем поддерживается редукция гипероксидирования пероксиредоксина [15]. Тиоредоксин — низкомолекулярный многофункциональный тиолсодержащий белковый фактор, SH-группы которого находятся либо в окисленном (SS-группа), либо в восстановленном состоянии (SH-группа) [16]. Именно в восстановленной форме (при сохраненных SH-группах) возможно ассоциирование его с редоксзависимой киназой ASK-1, которая является одним из основных факторов, запускающих программу гибели клеток, что сдерживает и даже ингибирует процесс апоптоза в целом [16] (рис. 1).
При инициировании синаптических NMDA-рецепторов важным компонентом целлюлярной выживаемости ткани мозга является активация фосфорилированием уже упомянутого ранее соединения CREB и включение сигналинга с участием последнего [12, 17, 18].
Помимо описанных выше процессов, как это ни парадоксально, не меньшее значение приобретает нуклеарный Са++ (ядерные ионы Са++), который в этом случае проявляет свойства триггерного компонента уже не повреждающих, а цитопротекторных процессов (рис. 1).
Доказано, что вход Са++ в ядро индуцирует активирование CREB и, соответственно, экспрессию генов, кодирующих фактор роста BDNF [12]. При этом отмечается вовлечение в описанный процесс кальциевых потенциалзависимых (или вольтзависимых) каналов L-типа. Поэтому при острых ситуациях ишемии/реперфузии мозга целесообразно тщательно продумывать вариант терапии с применением блокаторов кальциевых каналов, которые преимущественно блокируют потенциалзависимые каналы медленной проводимости L-типа.
Нельзя не отметить и факт высвобождения ионов Са++ из интрацеллюлярных кальциевых депо без влияния на уровень нуклеарного Са++, что в то же время, при активации синаптических NMDA-рецепторов, создает условия для активирования СREB-транскрипционных механизмов [19]. В связи с этим также отмечается модулирующее влияние нуклеарного Са++ на транскрипционно-зависимые процессы памяти и когнитивных функций. Значимым компонентом описанных процессов является механизм включения активирующего транскрипционного фактора Atf-3 (Аctivating transcription factor-3), индуцируемого активированным CREB под влиянием входящих ионов Са++ при возбуждении синаптических NMDA-рецепторов [20] (рис. 1).
Таким образом, имеет место функционирование сигналингового пути, когда запускающим компонентом является активация синаптических NMDA-рецепторов, а в эффекторной системе — нуклеарный Са++.
Логическое завершение такого сигналинга — активация геномной программы выживаемости, включая экспрессию генов клетки, кодирующих индукцию протекторных компонентов и поддерживающих адаптивные изменения, в свою очередь направленные на сохранение жизнедеятельности и подавление генного пула гибели [21–23]. В настоящее время продолжают оставаться актуальными и заслуживают внимания исследования по раскрытию механизма поддержания жизнедеятельности нейронов в условиях стрессорного влияния и/или депрессии аэробного метаболизма.
Геномная программа нейропротекции, ассоциированная с синаптической активностью NMDA-рецепторов, распространяется на пул генов, основу которого составляет комплекс, определяемый как активно-регулируемый ингибитор смерти — AID (activity-regulated inhibitor of death) [24].
Описанный механизм также лежит в основе блокирования и сдерживания процессов гибели клетки, включая условия развития эксайтотоксичности. Преимущество возбуждения синаптических NMDA-рецепторов состоит в активном контроле кальцийзависимой нейропротекторной транскрипционной программы. Формирующийся потенциал действия при этом супрессирует генкодируемый проапоптотический фактор р53 и угнетает регулирующую активность целевых генов, отвечающих за продукцию активирующих апоптоз факторов, в том числе и PUMA, а также апоптотических ферментов — прокаспазы-9 и каспазы-3 [25, 26] (рис. 1). С учетом описанной связи трансдукционных и транскрипционных факторов важно отметить, что это одна из сторон многокомпонентной системы адаптивных процессов нейронально-глиальной выживаемости, одним из логических завершений которой является стабилизация функционирования митохондрий и предупреждение развития митохондриально-зависимого апоптоза.
/26.jpg)
Таким образом, при ишемии/реперфузии ткани головного мозга благодаря активности NMDA-рецепторов синаптической локализации ингибируется комплекс геномного пула апоптотического ядерного фактора, что, в свою очередь, ведет к созданию условий для ингибирования запуска апоптотической программы и активации или поддержания процессов целлюлярной выживаемости.
Тем не менее имеются данные о значении экстрасинаптических NMDA-рецепторов в условиях, исключающих дефицит кислорода и депрессию аэробного метаболизма. Так, физиологическую роль активации экстрасинаптических NMDA-рецепторов связывают с функцией резерва для включения дополнительных синапсов [27], с возможностью проявления синаптической пластичности, расширением возможностей взаимодействия нейронов и глии, а также ретроградного сигналинга [28]. В физиологических условиях функционирование экстрасинаптической глутаматергической и ГАМКергической систем осуществляется на основе общих принципов нейротрансмиссии и взаимной диффузии сигналов [29].
Вышепредставленная информация свидетельствует о том, что при критическом состоянии головного мозга при сохранении двух противоположных эффектов функционирования NMDA-рецепторов в зависимости от синаптической и экстрасинаптической локализации в условиях острой ишемии/реперфузии доминирует экстрасинаптический. При этом, хотя неотъемлемым патогенетическим процессом является эксайтотоксичность с включением апоптотической программы, при поддержании выживаемости клеток следует учитывать ведущую роль синаптической активности NMDA-рецепторов. Это обусловливает совершенно новый терапевтический подход к построению нейропротективной стратегии. В таком случае, наряду с устранением дефицита кислорода и коррекцией энергетического метаболизма, необходимо предусматривать целесо–образность поддержания синаптической NMDA-рецепции, что в конечном счете создаст условия для выживаемости ткани мозга, а в отдаленном периоде уменьшит проявления когнитивного и неврологического дефицита.
Список литературы
1. Wiegert J.S., Bading H. Activity-dependent calium signaling and ERK-MAP kinases in neurons:a link to structural plasticity of the nucleus and gene transcription regulation // Cell Calcium. 2011 May; 49(5): 296-305.
2. Ivanov A., Pellegrino C., Rama S., Dumalska I., Salyha Y., Ben-Ari Y., Medina I. Opposing role of synaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons // J. Physiol. 2006, May 1; 572 (Pt 3): 789-89.
3. Krapivinsky G., Krapivinsky L., Manasian Y., Ivanov A., Tyzio R., Pellegrrino C., Ben-Ari Y., Clapham D.E., Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1 // Neuron. 2003, Nov 13; 40(4): 775-84.
4. Petralia R.S., Wang Y.X., Hua F., Yi Z., Stephenson F.A., Wenthold R.J. Organization of NMDA receptors at extrasynaptic location // Neuroscience. 2010, Apr 28; 167(1): 68-87.
5. Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders // Nat. Rev. Neurosci. 2010 Oct; 11(10): 682-96.
6. Кудряшов И.Е. Глутаматергические ионотропные рецепторы и потенциал-зависимые дендритные каналы в гиппокампе: их взаимодействие в пластических процессах // Нейрохимия. — 2003. — Т. 20, № 2. — С. 85-92.
7. Leveille F., Gaamouch F., Gouix E., Lecocg M., Lobner D., Nicole O., Buisson A. Neuronal viability is controlled by afunctional relation between synaptic and extrasynaptic NMDA receptors // FASEB J. 2008 Dec; 22(12): 4258-4271.
8. Dick O., Bading H. Synaptic activity and nuclear calcium signaling protect hippocampal neurons from death signal-associated nuclear translocation of FoxO3a induced by extrasynaptic N-methyl-D-aspartate receptors // J. Biol. Chem. 2010, Jun 18; 285(25): 19354-61.
9. Tu W., Xu X., Peng L., Zhong X., Zhang W., Soundarapandian M.M., Balel C., Wang M., Jia N., Zhang W., Lew F., Chan S.L., Chan Y., Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke // Cell. 2010, Jan 22; 140(2): 222-34.
10. Wahl A.S., Buchthal B., Rode F., Bomholt S.F., Freitag H.E., Hardingham G.E., Ronn L.C., Bading H. Hypoxic/ischemic conditions induce expression of the putative pro-death Clca1 via activation of extrasynaptic N-methyl-D-aspartate receptors // Neuroscience. 2009, Jan 12; 158(1): 344-52.
11. Zang S.J., Steijaert M.N., Lau D., Schutz G., Delucinge-Vivier C., Dascombes P., Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death // Neuron. 2007, Feb 15; 53(4): 549-62.
12. Hardingham G.E., Fukunaga Y., Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways // Nat. Neurosci. 2002 May; 5(5): 405-14.
13. Гусев Е.И., Скворцова В.И. Ишемия головного мозга. — М., 2001. — C. 326.
14. Al-Mubarak B., Soriano F.X., Hardingham G.E. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene // Channels (Austin). 2009 Jul-Aug; 3(4): 233-8.
15. Papadia S., Soriano F.X., Leveille F., Martel M.A., Dakin K.A., Hansen H.H., Kaindi A., Sifringer M., Fowler J., Stefovska V., McKenzie G., Craiqon M., Corriveau R., Ghazal P., Horsburqh K., Yankner B.A., Wyllie D.J., Ikonomidou C., Hardingham G.E. Synaptic NMDA receptor activity boots intrinsic antioxidant defenses // Nat. Neurosci. 2008 Apr: 11(4): 476-87.
16. Губский Ю.И., Беленичев И.Ф., Левицкий Е.Л., Горбачева С.В., Бухтиярова Н.В., Задорина О.В. Роль активных форм кислорода в функциональной активности MAP-киназного каскада, глобальных факторов транскрипции и развитии апоптоза (обзор литературы и собственных исследований) // Журн. АМН України. — 2008. — Т. 14, № 2. — С. 203-217.
17. Lee B., Butcher G.Q., Hoyt K.R., Impey S., Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein(CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133 // J. Neurosci. 2005, Feb 2; 25(5): 1137-48.
18. Hardingham G.E., Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is development regulated // Biochim. Biophys. Acta. 2002, Nov 4; 1600 (1–2): 148-153.
19. Hardingham G.E., Arnold F.J., Bading H. Nuclear calcium signaling control CREB- mediated gen expression triggered by synaptic activity // Nat. Neurosci. 2001 Mar; 4(3): 261-7.
20. Zhang S.J., Buchthal B., Lau D., Haver S., Dick O., Schwaninger M., Veltkamp R., Zou M., Weiss U., Bading H. A signal cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module theet protects agains extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage // J. Neurosci. 2011, Mar 30; 31(13): 4978-90.
21. Zhang S.J., Steijaert M.N., Lau D., Schutz G., Delucinge-Vivier C., Descombes P., Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death // Neuron. 2007, Feb 15; 53(4): 549-62.
22. Papadia S., Stevenson P., Hardingham N.R., Bading H., Hardingham G.E. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection // J. Neurosci. 2005, Apr 27; 25(17): 4279-87.
23. Hardingham G.E., Chawla S., Johnson C.M., Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression // Nature. 1997, Jan 16; 385(6613): 260-265.
24. Zhang S.J., Zou M., Lu L., Lau D., Ditzel D.A., Delucinge-Viver C., Aso Y., Descombes P., Bading H. Nuclear calcium signaling controls expression of large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity // PloS Genet. 2009 Aug; 5(8): e1000604.
25. Leveille F., Papadia S., Fricker M., Bell K.F., Soriano F.X., Wyllie D.J., Ikonomidou C., Tolkovsky A.M., Haredingham G.E. Suppression of the intrinsic apoptosis pathway by synaptic activity // J. Neurosci. 2010, Feb 17; 30(7): 2623-35.
26. Lui D., Bading H. Synaptic activity-mediated suppression of p53 and indication of nuclea calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition // J. Neurosci. 2009, Apr 8; 29 (14): 4420-29.
27. Zhou X., Hollern D., Liao J., Andrechek E., Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors // Cell Death Dis. 2013, Mar 28; 4: e560. doi: 10.1038/cddis.2013.82.
28. Vizi E.S., Fekete A., Karoly R., Mike A. Non-synaptic receptors and transporters involeved in brain function and targets of drug treatment // Br. J. Pharmacol. 2010 Jun; 160(4): 785-809.
29. Semyanov A.V. Diffusional extrosynaptic neurotransmission via glutamate and GABA // Neurosci. Behav. Physiol. 2005; 35(3): 253-366.