Введение
Данная статья является продолжением серии публикаций, посвященных нозоспецифическим особенностям антиоксидантной системы при заболеваниях органов дыхания.
Бронхиальная астма
Общепризнано, что существует связь между активностью генерации активированных кислородсодержащих метаболитов (АКМ), активированных азотсодержащих метаболитов (ААМ) и тяжестью течения бронхиальной астмы (БА). У больных БА высокий уровень генерации АКМ ассоциирован с выраженностью нейтрофильной инфильтрации слизистой оболочки бронхов и коррелирует как со степенью гиперреактивности бронхиального дерева, так и с риском летального исхода заболевания [4, 43]. У больных БА наблюдается усиление активности дуальной оксидазы 1 (dual oxidase 1 — DUOX1). Экспрессия фермента DUOX1 индуцируется –Тh2-ассоциированными интерлейкинами — IL-4 и -13, которые являются основными эффекторными цитокинами аллергических заболеваний респираторного тракта, в то время как экспрессия фермента DUOX2 индуцируется Тh1-ассоциированным цитокином IFN-γ [10]. Нарушение функционирования оксидаз NOX (NADPH-oxidase) ассоциировано с риском развития аллергениндуцированного воспаления дыхательных путей [13]. Для больных БА характерна гиперэкспрессия оксидазы NOX4 в гладкомышечных клетках. Повышенная активность NOX4 способствует развитию фиброза легочной ткани, а также пролиферации и гипертрофии гладкомышечных клеток бронхиального дерева, вероятно, предопределяя ремодулирование стенки бронхов [29, 41].
У больных БА достоверно повышается генерация АКМ и ААМ, и показано, что данная супергенерация свободных радикалов предопределяет развитие хронического воспалительного процесса респираторного тракта. При БА окисидантный стресс индуцирует продукцию различных провоспалительных медиаторов, способствует развитию гиперреактивности бронхов, стимулирует бронхоспазм и усиливает секрецию муцина [8, 52, 55].
Показано, что уже через 10 минут после взаимодействия аллергена с эпителиоцитами респираторного тракта отмечается не менее чем двукратное увеличение генерации супероксида анион-радикала (O2–•), которое сохраняется на всем протяжении аллергической реакции. Приступ БА непосредственно ассоциирован с генерацией O2–•, скорость которой приблизительно составляет 4 • 106 нмоль/5 • 105 клеток/ч. У больных с атопической формой БА в выдыхаемом воздухе идентифицируется концентрация NO в среднем в 3 раза выше, чем у здоровых людей. Повышенная генерация монооксида азота обусловлена активацией транскрипции гена iNOS эпителиоцитов респираторного и усилением катаболического распада S-нитрозоглутатиона (GSNO) как пула хранения NO в легочной ткани [5]. GSNO является физиологическим субстратом для глутатионзависимой формальдегид-дегидрогеназы (алкогольдегидрогеназы III класса), которая получила название GSNO-редуктазы (GSNOR). Редуктаза GSNOR, восстанавливая S-нитрозоглутатион, предупреждает нитрозилирование других белков.
У мышей с нокаутом гена Gsnor отмечается достоверное повышение уровня содержания SNO-протеинов в сочетании с низким уровнем чувствительности бронхиального дерева к бронхоконстрикторным агентам [48]. Мыши с мутацией данного гена характеризуются сочетанием низкого уровня риска развития инфаркта миокарда с высоким уровнем риска летального исхода при экспериментальном эндотоксическом шоке. Интерес вызывает тот факт, что назначение ингибиторов iNOS мышам с нокаутом гена Gsnor предупреждает неблагоприятный исход шока [19]. Бронхиальная астма сопровождается повышением содержания GSNOR в жидкости бронхоальвеолярного лаважа, и уровень концентрации GSNOR коррелирует с выраженностью бронхоконстрикции. L.G. Que и соавт. [54] считают, что GSNOR является важнейшим молекулярным фактором, участвующим в развитии гиперреактивности бронхиального дерева. Установлено, что SNP rs1154404 и rs28730619 гена Gsnor ассоциированы с высоким риском развития БА у детей [21].
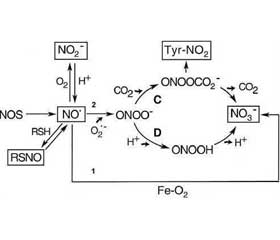
По сравнению с контролируемой БА более тяжелые ее формы сопровождаются значительным повышением уровня концентрации NO3– и нитротирозинов в выдыхаемом воздухе. Уже через несколько минут после индукции аллергеном у больных БА наблюдаются достоверное повышение концентрации NO3– и тенденциозное снижение концентрации NO• в выдыхаемом воздухе [40]. Уменьшение концентрации NO• вызвано, вероятно, реакцией NO• с O2–•, обусловливающей появление пероксинитрита, который в последующем распадается с образованием NO3– или участвует в формировании нитротирозинов. Содержание нитротирозинов у больных с тяжелой формой БА на порядок выше, чем у здоровых людей (рис. 1).
/729-1.jpg)
Течение БА сопряжено со снижением активности как супероксиддисмутазы (superoxide dismutase — SOD), так и каталазы в респираторном тракте. У больных БА вне зависимости от периода болезни в жидкости бронхоальвеолярного лаважа наблюдается низкая активность SOD, причем снижение ее активности особенно выражено во время обострения заболевания. Отмечено, что выявленное уменьшение уровня активности SOD при атопической форме БА происходит через несколько минут после контакта с причинно-значимым аллергеном. Снижение активности SOD сопровождается увеличением генерации O2–• эпителиоцитами респираторного тракта. Степень снижения активности SOD пропорциональна уровню гиперреактивности бронхиального дерева и тяжести заболевания [6, 35, 39]. Вероятно, что снижение активности дисмутирования супероксида анион-радикала связано с окислением и нитрованием молекулы ECSOD, так как дефицит активности дисмутирования в первую очередь отмечается в жидкости бронхоальвеолярного лаважа, в то время как активность CuZnSOD и MnSOD в сыворотке крови больных с легкой формой БА не изменяется. Снижение активности CuZnSOD и MnSOD происходит только при тяжелом течении заболевания. Также показано, что SNP Ala16Val гена MnSOD, возможно, ассоциирован с риском развития БА [57]. Однако, согласно данным других исследований, SNP Ala16Val не влияет на вероятность возникновения БА [50].
Установлено, что при БА, особенно у детей, происходит значительное (практически на 50 %) снижение активности каталазы как в жидкости бронхоальвеолярного лаважа, так и в эритроцитах периферической крови. По всей вероятности, низкий уровень активности каталазы играет важную патогенетическую роль в развитии аллергического процесса. Показано, что SNP 262C > T гена каталазы CAT ассоциирован с атопической формой БА [45]. Снижение активности каталазы не сопряжено с уменьшением молекулярного представительства данного фермента. По-видимому, снижение активности каталазы обусловлено нарушением соотношения хлорирования и нитрования тирозиновых остатков протеина каталазы [5]. Интересно, что у больных БА процессы хлорирования в 20 раз активнее, чем нитрования тирозиновых остатков протеинов. Таким образом, в органах дыхания у больных БА O2–•, генерируемый при респираторном взрыве, недостаточно димутируется SOD в перекись водорода, однако ее концентрация из-за дефицита активности каталазы в местах поражения респираторного тракта достигает достаточно высокого уровня. Снижение контроля над уровнем продукции H2O2 обусловливает усиление процессов окисления, нитрования, галогенирования протеинов и, как следствие, воспалительного процесса в респираторном тракте больных БА [39].
Характерной особенностью БА является высокая активность пероксидазы эозинофилов и миелопероксидазы в жидкости бронхоальвеолярного лаважа и сыворотке периферической крови. Высокая активность пероксидазы эозинофилов сопровождается появлением соединений брома, в частности 3-бромтирозина [5]. В последнее время в качестве тестов для неинвазивной диагностики и прогноза течения БА предлагается использовать определение экскреции бромтирозина с мочой и уровень концентрации монооксида азота в выдыхаемом воздухе [62].
У больных БА, кроме низкой активности SOD и каталазы, наблюдается пониженное содержание β-каротинов, аскорбиновой кислоты и токоферолов. Отличительным признаком БА можно считать повышение активности в жидкости бронхоальвеолярного лаважа и экспрессии глутатионпероксидаз (glutathione peroxidase — GPX) — eGPX/GPX3, GPX2 и глутатион-S-трансферазы Omega 1-1 (glutathione S-transferase omega 1 — GSTO1). Установлено, что повышение активности данных ферментов, наблюдаемое при БА, определяет развитие аллергического воспаления дыхательных путей [34, 42]. Продемонстрировано, что SNP Ile105Val (rs1695) гена GSTP1 (glutathione S-transferase pi 1) ассоциирован с развитием БА [20, 26]. Также показано, что полиморфизмы генов GSTP1, GSTM1, GSTT1 у матери являются факторами, несущими риск развития БА у ее детей (Wu C.C. и соавт., 2012). Увеличение генерации АКМ и снижение активности GR, наблюдаемые при БА, закономерно сопровождаются снижением уровня глутатиона и повышением соотношения GSSG/GSH в респираторном тракте. У детей, больных БА, во время приступа значительно ниже уровень концентрации глутатиона в выдыхаемом воздухе, чем у детей контрольной группы. Обострение БА сопровождается быстрым изменением как внутриклеточного, так и внеклеточного соотношения GSSG/GSH — уже через несколько минут после воздействия аллергена происходит снижение концентрации GSH и повышение содержания GSSG в надэпителиальной жидкости респираторного тракта [5, 51]. Считают, что повышение концентрации GSSG, наблюдаемое при БА, обусловлено увеличением активности гистоновых ацетилтрансфераз, подавлением активности гистоновых деацетилаз в клетках респираторного тракта. Повышение концентрации GSSG сопровождается усилением продукции провоспалительных цитокинов, в том числе и IL-8/CXCL8, рекрутирующего нейтрофилы [44], и развитием макрофагальной дисфункции [24]. Дефицит GSH обусловливает ингибирование продукции Th1-ассоциированных цитокинов и активацию их синтеза [51], в то время как увеличение содержания GSH в клетках респираторного тракта приводит к снижению содержания IL-4, -5, -10, эотаксина, CCL5/RANTES в жидкости бронхоальвеолярного лаважа и усилению продукции IL-12, IFN-γ [25].
При БА, особенно во время обострения, наблюдается высокий уровень экспрессии тиоредоксинов (thioredoxin — TRX) и пероксиредоксинов (peroxiredoxin — PRX), а гипероксидация последних коррелирует с тяжестью заболевания [14, 27]. Относительно высокий уровень TRX и PRX, по всей вероятности, играет протективную роль. Так, TRX1 индуцирует экспрессию Th1-ассоциированных цитокинов, обусловливает снижение реактивности и степени воспаления бронхиального дерева, предотвращает развитие гиперплазии бокаловидных клеток, ингибируя экспрессию генов MUC и Gob-5 [11, 60, 61]. TRX ингибируют продукцию фактора, подав–ляющего макрофагальную миграцию (macrophage migration inhibitory factor), и эотаксина, тем самым ограничивая рекрутирование эозинофилов в слизистую оболочку бронхиальной стенки [59]. Показано, что PRX1, ингибируя продукцию IL-2, снижает активность Th2-ассоциированного воспалительного процесса в респираторном тракте [46]. Таким образом, редокс-процессы поддерживают Th2-ассоциированный ответ, характерный для БА (рис. 2).
/730-1.jpg)
Хроническая обструктивная болезнь легких
При хронической обструктивной болезни легких (ХОБЛ), как и при БА, наблюдается повышенное образование АКМ и ААМ, избыток которых сопровождается окислением и нитрованием протеинов и, возможно, деструкцией клеток. Особый вклад в генерацию АКМ у больных ХОБЛ привносят альвеолярные макрофаги и рекрутированные нейтрофилы. В то же время у больных ХОБЛ наблюдается снижение экспрессии DUOX1 и DUOX2 эпителиоцитами респираторного тракта. Представляет интерес то, что у курильщиков отмечается снижение экспрессии только DUOX1. АКМ индуцируют воспаление, апоптоз клеток, активируют протеазы и инактивируют антипротеазы в тканях органов дыхания. Концентрации супероксида радикал-аниона и перекиси водорода, которые генерируются эпителиоцитами и фагоцитами альвеол легких больных ХОБЛ, достаточны, чтобы инактивировать основной человеческий лейкоцитарный ингибитор эластазы — α1-антитрипсин, играющий ключевую роль в предотвращении развития эмфиземы. У больных ХОБЛ отмечается повышение уровня 8-гидрокси-2'-деоксигуанозина в моче и сыворотке крови и Н2О2, изопростана F2α-III, нитротирозинов в выдыхаемом воздухе. Уровень концентрации данных веществ коррелирует с тяжестью заболевания [33, 49]. Однако АКМ играют неоднозначную роль в патогенезе ХОБЛ. Исследование влияния сигаретного дыма на респираторный тракт мышей с нокаутом генов p47phox и gp91phox показало, что отсутствие активности НАДФH-оксидазы сопровождается усилением продукции провоспалительных цитокинов (CCL2/МСР-1, IL-6, TNF-α и/или GM-CSF) и выраженности воспалительного процесса ткани легкого [22].
В проведенных исследованиях показано, что в легочных макрофагах и в клетках ткани легкого пациентов с ХОБЛ отмечается подавление активности фактора транскрипции NRF2 (nuclear factor, erythroid 2 like 2) посттрансляционными модификациями протеина KEAP1 (kelch like ECH associated protein 1) [9, 47].
Патогенетической особенностью оксидантного стресса, наблюдаемого при ХОБЛ, является существенный вклад железозависимых окислительно-восстановительных реакций. Данный эффект обусловлен индуцированным сигаретным дымом с привлечением макрофагов, нейтрофилов, повышенной экспрессией железорегуляторного протеина 2 (IREB2 или IRP2) и увеличением концентрации железосодержащих молекул (трансферрина и лактоферрина) в ткани легкого у больных ХОБЛ. Установлено, что протеолиз трансферрина и лактоферрина приводит к увеличению содержания свободного железа в надэпителиальной жидкости и межклеточном пространстве. Железозависимые окислительно-восстановительные реакции приводят к появлению высокореактивных гидропероксидного (H2O2 + Fe3+ → Fe2+ + OOH• + H) и гидроксильного (H2O2 + Fe2+ → Fe3+ + OH• + OH–) радикалов [18, 30].
Как и у больных БА, так и у пациентов с ХОБЛ наблюдается повышенная активность миелопероксидазы нейтрофилов, уровень активности которой сопряжен со степенью нарушения вентиляции легких. Представляет научный интерес тот факт, что у больных с ХОБЛ, несмотря на рекрутирование эозинофилов в регионы воспаления респираторного тракта, не наблюдаются повышение активности пероксидазы эозинофилов и образование 3-бромтирозина [49].
Повышение концентрации NO в выдыхаемом воздухе характерно только для периода обострения ХОБЛ [63]. У больных ХОБЛ (особенно курильщиков) характерно наличие высоких уровней концентрации пероксинитрита (ONOO–), нитрованых белков (фибриноген, трансферрин, плазминоген, церулоплазмин) [49].
У больных с ХОБЛ наблюдается снижение экспрессии фактора транскрипции NRF2 при одновременном повышении содержания протеинов KEAP1 и BACH1 (BTB domain and CNC homolog 1) в ткани легкого. Продемонстрировано, что чем ниже уровень экспрессии NRF2, тем тяжелее проявления эмфиземы легкого. Дефицит представительства NRF2 сопровождается снижением экспрессии его генов-мишеней, которые кодируют антиоксидантные протеины гемоксидазы, НАДФН-дегидрогеназу хинон-1 и глутатионпероксидазу-2 в альвеолярных макрофагах больных с эмфиземой легкого. Скорее всего, высокая экспрессия протеина KEAP1, способствуя протеасомной деградации NRF2, обусловливает его дефицит и, как следствие, низкую активность антиоксидантной системы [1, 7].
A. Boutten и соавт. [47] считают, что медикаментозная модуляция активности NRF2-ассоциированного сигнального пути может стать важным терапевтическим подходом для профилактики эмфиземы легких у больных с ХОБЛ.
У больных с ХОБЛ наблюдается низкая активность ECSOD в жидкости бронхоальвеолярной жидкости, которая сопряжена с индукцией эластазы нейтрофилов и развитием эмфизематоза легких [16, 17]. Применение SOD-миметических средств или SOD-содержащих препаратов препятствует развитию эмфиземы легких [58]. Установлено, что у людей с полиморфизмом R213G гена ECSOD, который сопровождается очень высоким содержанием ECSOD (ее уровень в 10–20 раз выше, чем у людей без данного полиморфизма) в жидкости бронхоальвеолярного лаважа и сыворотке крови, достоверно реже развивается ХОБЛ. Высокий уровень концентрации ECSOD в биологических жидкостях у людей с полиморфизмом R213G гена ECSOD обусловлен потерей способности ее молекулы связываться с поверхностью мембраны клеток [23]. Однако другие SNP гена ECSOD — rs8192287, rs9192288, которые сопровождаются снижением функциональной активности фермента ECSOD, ассоциированы со снижением функции легких и высоким риском развития эмфиземы легких [56].
При изучении активности экспрессии 42 генов кандидатов установлено, что при ХОБЛ отмечается достоверное снижение содержания мРНК ECSOD, каталазы, GSTP1, GSTM1, микросомальной эпоксид-гидролазы (epoxide hydrolase 1 — mEPHX) и тканевого ингибитора металлопротеиназы-2 (metallopeptidase inhibitor 2 — TIMP2), степень снижения которых коррелирует со степенью нарушения вентиляции легких. Дефицит активности антиоксидантной системы при ХОБЛ сопряжен с увеличением продукции провоспалительных цитокинов: IL-1β, -6, -8/CXCL8, CCL2/МСР-1, CCL8/MCP-2, CXCL1/Gro-α [16, 38, 53].
У больных с ХОБЛ отмечается напряжение процесса синтеза глутатиона, которое характеризуется повышением активности γ-глутамилтрансферазы в эпителии респираторного тракта [12, 36]. Показано, что уровень GSH в бронхоальвеолярной жидкости зависит от стажа курения. Первоначально отмечается значительное повышение концентрации GSH, а в последующем происходит постепенное ее снижение. Одним из наиболее интересных аспектов является то, что наблюдаемое снижение концентрации GSH в бронхоальвеолярной жидкости сопряжено со снижением его содержания в сыворотке крови и печени, в то время как уровень GSH в ткани легкого остается неизменным. По всей вероятности, ткань легкого контрибутирует глутатион из сыворотки крови. Основным источником GSH в организме человека является печень, а оксидантный стресс при заболеваниях органов дыхания сопряжен с повышением активности γ-глутамилтрансферазы (gamma-glutamyltransferase — GGT) в гепатоцитах [32]. Также одним из важнейших компонентом системы, регулирующей уровень концентрации GSH, является окись углерода (СО), которая индуцирует активность глутаматцистеинлигазы (glutamate-cysteine ligase — GCL) [3]. Снижение уровня концентрации GSH способствует продукции провоспалительных цитокинов.
Установлено, что при ХОБЛ отмечается усиление активности глутатионпероксидаз (GPX1, GPX2, GPX3) и глутатионредуктазы в эпителиальных клетках и альвеолярных макрофагах респираторного тракта, особенно курильщиков, в то время как экспрессия тиоредоксина и активность тиоредоксинредуктазы практически не изменяются. Длительно текущие ХОБЛ сопровождаются изменениями в активности пероксиредоксинов — усилением активности PRX1 и PRX3 и ингибированием экспрессии PRX5 [2, 15].
Табачный дым содержит более чем 5000 различных химических соединений и примерно 1017 молекул свободных радикалов в объеме одной затяжки сигареты. Компоненты табачного дыма и другие ирританты, попадающие в легкие во время акта дыхания, привлекают макрофаги и активируют как рекрутируемые, так и альвеолярные макрофаги [49]. Различные экзогенные триггерные факторы отличаются некоторыми особенностями вызываемых эффектов (табл. 1).
Активация макрофагов приводит к выраженной продукции АКМ, ААМ и хемокинов, привлекающих другие эффекторные и регулирующие клетки (рис. 3). Повышение уровня генерации АКМ инициирует воспалительную реакцию в легких, активируя факторы транскрипции NF-κB, AP-1, определяющие экспрессию генов провоспалительных медиаторов.
/732-1.jpg)
Хроническое действие табачного дыма обусловливает накопление макрофагов, нейтрофилов и CD8+ Т-клеток в легочной ткани. Макрофаги и нейтрофилы высвобождают различные протеазы, в том числе нейтрофильную эластазу, протеиназу-3, матриксные металлопротеиназы (matrix metallopeptidase — MMP), катепсины. АКМ, в свою очередь, активируют латентные проформы MMP, которые вместе с другими протеиназами, расщепляя компоненты экстрацеллюлярного матрикса, нарушают его целостность, а образованные фрагменты эластина и коллагена, например пролин-глицин-пролина, усиливают хемотаксис клеток-предшественников макрофагов и нейтрофилов. Нарушение структуры экстрацеллюлярного матрикса лежит в основе развития эмфизематоза легких. Значение АКМ в развитии ХОБЛ также заключается в том, что назначение антиоксидантов (N-ацетилцистеина, пирролидиндитиокарбамата), ингибиторов НАДФH-оксидазы (дифенилен иодония хлорида и апоцинина) предупреждает развитие эмфизематоза легких [18, 31].
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Goven D, Boutten A, Leçon-Malas V, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008 Oct;63(10):916-24. doi: 10.1136/thx.2007.091181. Epub 2008 Jun 17.
2. Bentley AR, Emrani P, Cassano PA. Genetic variation and gene expression in antioxidant related enzymes and risk of COPD: a systematic review. Thorax. 2008 Nov;63(11):956-61. doi: 10.1136/thx.2007.086199. Epub 2008 Jun 19.
3. Li MH, Jang JH, Na HK, Cha YN, Surh YJ. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress inducesexpression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J Biol Chem. 2007 Sep 28;282(39):28577-86. Epub 2007 Aug 5. doi: 10.1074/jbc.M701916200.
4. Cho YS, Moon HB. The role of oxidative stress in the pathogenesis of asthma. Allergy Asthm Immunol Res. 2010 Jul;2(3):183-7. doi: 10.4168/aair.2010.2.3.183. Epub 2010 Apr 29.
5. Comhair SA, Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2010 Jan;12(1):93-124. doi: 10.1089/ARS.2008.2425.
6. Comhair SA, Ricci KS, Arroliga M, et al. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med. 2005 Aug 1;172(3):306-13. Epub 2005 May 5. doi: 10.1164/rccm.200502-180OC.
7. Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008 Sep 15;178(6):592-604. doi: 10.1164/rccm.200803-380OC. Epub 2008 Jun 12.
8. Dozor AJ. The role of oxidative stress in the pathogenesis and treatment of asthma. Ann N Y Acad Sci. 2010 Aug;1203:133-7. doi: 10.1111/j.1749-6632.2010.05562.x. doi:10.1111/j.1749-6632.2010.05562.x.
9. Suzuki M, Betsuyaku T, Ito Y, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2008 Dec;39(6):673-82. doi: 10.1165/rcmb.2007-0424OC. Epub 2008 Jun 19.
10. Nagai K, Betsuyaku T, Suzuki M, et al. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderatechronic obstructive pulmonary disease. Antioxid Redox Signal. 2008 Apr;10(4):705-14. doi: 10.1089/ars.2007.1941.
11. Imaoka H, Hoshino T, Okamoto M, et al. Endogenous and exogenous thioredoxin 1 prevents goblet cell hyperplasia in a chronic antigen exposure asthma model. Allergol Int. 2009 Sep;58(3):403-10. doi: 10.2332/allergolint.09-OA-0086. Epub 2009 Jun 25.
12. Cepelak I, Dodig S, Romic D, Ruljancic N, Popovic-Grle S, Malic A. Enzyme catalytic activities in chronic obstructive pulmonary disease. Arch Med Res. 2006 Jul;37(5):624-9. doi: 10.1016/j.arcmed.2006.01.004.
13. Won HY, Jang EJ, Min HJ, Hwang ES. Enhancement of Allergen-induced Airway Inflammation by NOX2 Deficiency. Immune Netw. 2011 Jun;11(3):169-74. doi: 10.4110/in.2011.11.3.169. Epub 2011 Jun 30.
14. Yamada Y, Nakamura H, Adachi T, et al. Elevated serum levels of thioredoxin in patients with acute exacerbation of asthma. Immunol Lett. 2003 Apr 3;86(2):199-205. PMID: 12644323.
15. Pierrou S, Broberg P, O'Donnell RA, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007 Mar 15;175(6):577-86. Epub 2006 Dec 7. doi: 10.1164/rccm.200607-931OC.
16. Yao H, Arunachalam G, Hwang JW, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15571-6. doi: 10.1073/pnas.1007625107. Epub 2010 Aug 16.
17. Oberley-Deegan RE, Regan EA, Kinnula VL, Crapo JD. Extracellular superoxide dismutase and risk of COPD. COPD. 2009 Aug;6(4):307-12. PMID: 19811392.
18. Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413-21. doi: 10.2147/COPD.S10770. Epub 2011 Aug 5.
19. Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009 Sep;15(9):391-404. doi: 10.1016/j.molmed.2009.06.007. Epub 2009 Aug 31.
20. Slager RE, Hawkins GA, Li X, Postma DS, Meyers DA, Bleecker ER. Genetics of asthma susceptibility and severity. Clin Chest Med. 2012 Sep;33(3):431-43. doi: 10.1016/j.ccm.2012.05.005. Epub 2012 Jul 7.
21. Wu H, Romieu I, Sienra-Monge JJ, Estela Del Rio-Navarro B, et al. Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. J Allergy Clin Immunol. 2007 Aug;120(2):322-8. Epub 2007 Jun 1. doi: 10.1016/j.jaci.2007.04.022.
22. Yao H, Edirisinghe I, Yang SR, et al. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am J Pathol. 2008 May;172(5):1222-37. doi: 10.2353/ajpath.2008.070765. Epub 2008 Apr 10.
23. Juul K, Tybjaerg-Hansen A, Marklund S, Lange P, Nordestgaard BG. Genetically increased antioxidative protection and decreased chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006 Apr 15;173(8):858-64. Epub 2006 Jan 6. doi: 10.1164/rccm.200509-1387OC.
24. Fitzpatrick AM, Teague WG, Burwell L, Brown MS, Brown LA; NIH/NHLBI Severe Asthma Research Program. Glutathione oxidation is associated with airway macrophage functional impairment in children with severe asthma. Pediatr Res. 2011 Feb;69(2):154-9. doi: 10.1203/PDR.0b013e3182026370.
25. Koike Y, Hisada T, Utsugi M, et al. Glutathione redox regulates airway hyperresponsiveness and airway inflammation in mice. Am J Respir Cell Mol Biol. 2007 Sep;37(3):322-9. Epub 2007 May 16. doi: 10.1165/rcmb.2006-0423OC.
26. Gu ML, Zhao J. Mapping and localization of susceptible genes in asthma. Chin Med J (Engl). 2011 Jan;124(1):132-43. PMID: 21362321.
27. Kwon HS, Bae YJ, Moon KA, et al. Hyperoxidized peroxiredoxins in peripheral blood mononuclear cells of asthma patients is associated with asthma severity. Life Sci. 2012 Apr 9;90(13-14):502-8. doi: 10.1016/j.lfs.2012.01.003. Epub 2012 Jan 20.
28. Tubby C, Harrison T, Todd I, Fairclough L. Immunological basis of reversible and fixed airways disease. Clin Sci (Lond). 2011 Oct;121(7):285-96. doi: 10.1042/CS20110062.
29. Sutcliffe A, Hollins F, Gomez E, et al. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsicairway smooth muscle hypercontractility in asthma. Am J Respir Crit Care Med. 2012 Feb 1;185(3):267-74. doi: 10.1164/rccm.201107-1281OC. Epub 2011 Nov 22.
30. DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009 Oct;85(4):493-502. doi: 10.1016/j.ajhg.2009.09.004.
31. Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012 Sep 1;84(5):581-90. doi: 10.1016/j.bcp.2012.05.005. Epub 2012 May 12.
32. Gould NS, Min E, Gauthier S, Martin RJ, Day BJ. Lung glutathione adaptive responses to cigarette smoke exposure. Respir Res. 2011 Oct 7;12:133. doi: 10.1186/1465-9921-12-133.
33. MacNee W. Oxidants and COPD. Curr Drug Targets Inflamm Allergy. 2005 Dec;4(6):627-41. PMID: 17305519.
34. Meyer HA, Dittrich AM, Hamelmann E. Different isoforms of glutathione peroxidase cause opposing effects during the development of allergic asthma in mice. Antioxid Redox Signal. 2011 Jan 1;14(1):169-70; author reply 170-1. doi: 10.1089/ars.2010.3591. Epub 2010 Nov 5.
35. Misso NL, Thompson PJ. Oxidative stress and antioxidant deficiencies in asthma: potential modification by diet. Redox Rep. 2005;10(5):247-55. doi: 10.1179/135100005X70233.
36. Mistry D, Stockley RA. Gamma-glutamyl transferase: the silent partner? COPD. 2010 Aug;7(4):285-90. doi: 10.3109/15412555.2010.496819.
37. Maes T, Provoost S, Lanckacker EA, et al. Mouse models to unravel the role of inhaled pollutants on allergic sensitization and airway inflammation. Respir Res. 2010 Jan 21;11:7. doi: 10.1186/1465-9921-11-7.
38. Nakamura H. Genetics of COPD. Allergol Int. 2011 Sep;60(3):253-8. doi: 10.2332/allergolint.11-RAI-0326.
39. Ghosh S, Janocha AJ, Aronica MA, et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J Immunol. 2006 May 1;176(9):5587-97. PMID: 16622028.
40. Dweik RA, Comhair SA, Gaston B, et al. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc Natl Acad Sci U S A. 2001 Feb 27;98(5):2622-7. Epub 2001 Feb 20. doi: 10.1073/pnas.051629498.
41. Sturrock A, Huecksteadt TP, Norman K, et al. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007 Jun;292(6):L1543-55. Epub 2007 Mar 16. doi: 10.1152/ajplung.00430.2006.
42. Guo CH , Liu PJ, Lin KP, Chen PC. Nutritional supplement therapy improves oxidative stress, immune response, pulmonary function, and quality of life in allergic asthma patients: an open-label pilot study. Altern Med Rev. 2012 Mar;17(1):42-56. PMID: 22502622.
43. Di Stefano A, Gnemmi I, Vicari C, Balbi B. Oxidant-antioxidant balance in asthma and the potential of antioxidant therapies. Monaldi Arch Chest Dis. 2010 Sep;73(3):96-8. doi:10.4081/monaldi.2010.292.
44. Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002 May-Jun;234-235(1-2):239-48. PMID: 12162440.
45. Islam T, McConnell R, Gauderman WJ, Avol E, Peters JM, Gilliland FD. Ozone, oxidant defense genes, and risk of asthma during adolescence. Am J Respir Crit Care Med. 2008 Feb 15;177(4):388-95. Epub 2007 Nov 29. doi: 10.1164/rccm.200706-863OC.
46. Inoue K, Takano H, Koike E, et al. Peroxiredoxin I is a negative regulator of Th2-dominant allergic asthma. Int Immunopharmacol. 2009 Oct;9(11):1281-8. doi: 10.1016/j.intimp.2009.07.010. Epub 2009 Aug 5.
47. Boutten A, Goven D, Artaud-Macari E, Bonay M. Protective role of Nrf2 in the lungs against oxidative airway diseases. Med Sci (Paris). 2011 Nov;27(11):966-72. doi: 10.1051/medsci/20112711012. Epub 2011 Nov 30. (in French).
48. Que LG, Liu L, Yan Y, et al. Protection from experimental asthma by an endogenous bronchodilator. Science. 2005 Jun 10;308(5728):1618-21. Epub 2005 May 26. doi: 10.1126/science.1108228.
49. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006 Jul;28(1):219-42. doi: 10.1183/09031936.06.00053805.
50. Crawford A, Fassett RG, Geraghty DP, et al. Relationships between single nucleotide polymorphisms of antioxidant enzymes and disease. Gene. 2012 Jun 15;501(2):89-103. doi: 10.1016/j.gene.2012.04.011. Epub 2012 Apr 14.
51. Reynaert NL. Glutathione biochemistry in asthma. Biochim Biophys Acta. 2011 Nov;1810(11):1045-51. doi: 10.1016/j.bbagen.2011.01.010. Epub 2011 Jan 31.
52. Riedl MA, Nel AE. Importance of oxidative stress in the pathogenesis and treatment of asthma. Curr Opin Allergy Clin Immunol. 2008 Feb;8(1):49-56. doi: 10.1097/ACI.0b013e3282f3d913.
53. Llinàs L, Peinado VI, Ramon Goñi J, et al. Similar gene expression profiles in smokers and patients with moderate COPD. Pulm Pharmacol Ther. 2011 Feb;24(1):32-41. doi: 10.1016/j.pupt.2010.10.010. Epub 2010 Oct 21.
54. Que LG, Yang Z, Stamler JS, Lugogo NL, Kraft M. S-nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med. 2009 Aug 1;180(3):226-31. doi: 10.1164/rccm.200901-0158OC. Epub 2009 Apr 24.
55. Sugiura H, Ichinose M. Oxidative and nitrative stress in bronchial asthma. Antioxid Redox Signal. 2008 Apr;10(4):785-97. doi: 10.1089/ars.2007.1937.
56. Dahl M, Bowler RP, Juul K, Crapo JD, Levy S, Nordestgaard BG. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Am J Respir Crit Care Med. 2008 Nov 1;178(9):906-12. doi: 10.1164/rccm.200804-549OC. Epub 2008 Aug 14.
57. Yang LL, Huang MS, Huang CC, et al. The association between adult asthma and superoxide dismutase and catalase gene activity. Int Arch Allergy Immunol. 2011;156(4):373-80. doi: 10.1159/000324448. Epub 2011 Aug 9.
58. Tanaka K, Tanaka Y, Miyazaki Y, et al. Therapeutic effect of lecithinized superoxide dismutase on pulmonary emphysema. J Pharmacol Exp Ther. 2011 Sep;338(3):810-8. doi: 10.1124/jpet.111.179051. Epub 2011 Jun 10.
59. Torii M, Wang L, Ma N, et al. Thioredoxin suppresses airway inflammation independently of systemic Th1/Th2 immune modulation. Eur J Immunol. 2010 Mar;40(3):787-96. doi: 10.1002/eji.200939724.
60. Ichiki H, Hoshino T, Kinoshita T, et al. Thioredoxin suppresses airway hyperresponsiveness and airway inflammation in asthma. Biochem Biophys Res Commun. 2005 Sep 9;334(4):1141-8. doi: 10.1016/j.bbrc.2005.07.007.
61. Ito W, Kobayashi N, Takeda M, et al. Thioredoxin in allergic inflammation. Int Arch Allergy Immunol. 2011;155 Suppl 1:142-6. doi: 10.1159/000327501. Epub 2011 Jun 1.
62. Wedes SH, Wu W, Comhair SA, et al. Urinary bromotyrosine measures asthma control and predicts asthma exacerbations in children. J Pediatr. 2011 Aug;159(2):248-55.e1. doi: 10.1016/j.jpeds.2011.01.029. Epub 2011 Mar 10.
63. Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011 Jul 15;254(2):72-85. doi: 10.1016/j.taap.2009.10.022. Epub 2011 Feb 4.

/729-1.jpg)
/730-1.jpg)
/733-1.jpg)
/732-1.jpg)