
Международный эндокринологический журнал Том 15, №1, 2019
Вернуться к номеру
Синдром Мак-Кьюна — Олбрайта — Брайцева (огляд літератури та клінічний випадок)
Авторы: Сорокман Т.В., Макарова О.В., Попелюк Н.О.
ВДНЗ України «Буковинський державний медичний університет», м. Чернівці, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
У статті наведені результати огляду літератури щодо синдрому Мак-Кьюна — Олбрайта — Брайцева й описаний клінічний випадок даного синдрому. Синдром Мак-Кьюна — Олбрайта — Брайцева (МОБ) — генетично зумовлене захворювання, що характеризується тріадою симптомів: наявністю специфічних плям кольору кави з молоком, фіброзною дисплазією кісток і різноманітними ендокринопатіями, найчастішою з яких є передчасний статевий розвиток. Частота зустрічальності захворювання у світі перебуває в межах від 1 випадку на 100 тисяч до 1 випадку на мільйон у загальній популяції. Синдром Мак-Кьюна — Олбрайта — Брайцева викликається мутацією в гені, відомому як GNAS1. Даний ген кодує альфа-субодиницю гуанідинтрифосфатзв’язуючого білка (G-білка), що стимулює утворення циклічного аденозинмонофосфату. Мутантний білок постійно активує аденілатциклазу, що призводить до мимовільного «включення» секреції гормонів. GNAS1 знаходиться на довгому плечі 20-ї хромосоми. Дана клітинна мутація відбувається на ранніх стадіях ембріогенезу. Описаний клінічний випадок викликає безперечний інтерес із точки зору дещо запізнілої діагностики синдрому, хоча патологічні переломи є класичними ознаками хвороби. Успіх лікування цього захворювання безпосередньо залежить від ранньої діагностики та своєчасного направлення дитини до відповідної ендокринологічної установи.
В статье приведены результаты обзора литературы, касающиеся синдрома Мак-Кьюна — Олбрайта — Брайцева, и описан клинический случай данного синдрома. Синдром Мак-Кьюна — Олбрайта — Брайцева — это генетически обусловленное заболевание, как правило, характеризующееся триадой симптомов: наличием специфических пятен цвета кофе с молоком, фиброзной дисплазией костей и разнообразными эндокринопатиями, наиболее частой из которых является преждевременное половое развитие. Частота встречаемости заболевания в мире варьирует от 1 случая на 100 тысяч до 1 случая на миллион в общей популяции. Синдром Мак-Кьюна — Олбрайта — Брайцева вызывается мутацией в гене, известном как GNAS1. Данный ген кодирует альфа-субъединицу гуанидинтрифосфатсвязывающего белка (G-белка), стимулирующего образование циклического аденозинмонофосфата. Мутантный белок постоянно активирует аденилатциклазу, что приводит к самопроизвольному «включению» секреции гормонов. GNAS1 находится на длинном плече 20-й хромосомы. Данная клеточная мутация происходит на ранних стадиях эмбриогенеза. Описанный клинический случай представляет несомненный интерес с точки зрения несколько запоздалой диагностики синдрома, хотя патологические переломы являются классическими признаками болезни. Успех лечения данного заболевания напрямую зависит от ранней диагностики и своевременного направления ребенка в соответствующее эндокринологическое учреждение.
The paper presents the results of literature review on McCune-Albright-Braytsev syndrome and describes the clinical case of this syndrome. McCune-Albright-Braytsev syndrome is a genetically determined disease, usually characterized by a triad of symptoms: the presence of specific cafe-au-lait spots, fibrous dysplasia of the bones and various endocrinopathies, the most frequent of which is premature sexual development. The incidence of this disease in the world varies from 1 case per 100,000 to 1 case per million in the general population. McCune-Albright-Braytsev syndrome is caused by a mutation in GNAS1 gene. This gene encodes the alpha subunit of guanosine triphosphate binding protein (G protein), which stimulates the formation of cyclic adenosine monophosphate (cAMP) that, among other things, regulates the work of many endocrine glands in the body. The mutant protein constantly activates adenylate cyclase, the intracellular level of cAMP increases, which leads to spontaneous “inclusion” of hormone secretion. GNAS1 is on the long shoulder of chromosome 20. This cell mutation occurs in the early stages of embryogenesis. The described clinical case is of unquestionable interest in terms of somewhat delayed diagnosis of the syndrome, although pathological fractures are the classic signs of the disease. The success of the treatment of this disease directly depends on the early diagnosis and timely referral of the child to the appropriate endocrinological institution.
синдром Мак-Кьюна — Олбрайта — Брайцева; етіологія; клініка; огляд
синдром Мак-Кьюна — Олбрайта — Брайцева; этиология; клиника; обзор
McCune-Albright-Braytsev syndrome; etiology; clinical presentation; review
Вступ
Синдром Мак-Кьюна — Олбрайта — Брайцева (МОБ, МКХ-10: Q78.1. — поліостозна фіброзна дисплазія; OMIM 174800: фіброзна дисплазія, fibrous dysplasia/McCune-Albright syndrome, FD/MAS) — рідкісне спорадичне захворювання, що характеризується дисплазією фіброзної кістки, плямами на шкірі і мінливими гіперфункціональними ендокринопатіями. Це панетнічне захворювання, передбачувана частота якого перебуває в межах від 1 : 100 000 до 1 : 1000 [1]. До цього захворювання схильні обидві статі, проте в дівчаток воно трапляється удвічі частіше, ніж у хлопчиків. Деякі автори дотримуються думки, що від синдрому страждають тільки дівчатка. Це може бути пояснено тим, що один із найбільш характерних симптомів захворювання — передчасне статеве дозрівання — у дівчаток відзначається в 9 разів частіше, ніж у хлопчиків. Інші прояви синдрому в представників обох статей трапляються приблизно з однаковою частотою [2].
Історія відкриття синдрому
Уперше в 1928 році російський хірург В.Р. Брайцев описав симптомокомплекс під назвою «фіброзні пухлини» (автор використав термін osteodystrophia fibrosa localisata cystica) [3]. У 1936 р. Донован Джордж МакКьюн (Donovan George McCune) опублікував статтю з описом дівчинки, у якої периферичний передчасний статевий розвиток поєднувався з наявністю вогнищ гіперпігментації, фіброзно-кістозними змінами кісткової тканини і гіпертиреозом [4]. Спочатку автор припускав, що причина захворювання — нейрофіброматоз, але вже через рік він опублікував ще один подібний випадок з описом випадків інших авторів, які виявили 9 пацієнтів із тією ж тріадою ознак у вигляді гіперпігментації, пошкодження кісткової тканини за типом фіброзно-кістозних вогнищ і передчасного статевого розвитку і 5 пацієнтів із двома ознаками з трьох [5]. Одночасно в тому ж році американський ендокринолог Фуллер Олбрайт (Albright Fuller) із колегами опублікував опис п’яти власних спостережень пацієнтів із тріадою ознак і також навів опис раніше опублікованих подібних випадків [6]. Докладний аналіз всіх відомих на той момент випадків дозволив Олбрайту висунути припущення про існування окремого захворювання, зумовленого дефектами ембріонального розвитку, і виділити його провідні ознаки, що лягли в основу критеріїв діагностики: плями кольору кави з молоком, периферичний передчасний статевий розвиток і характерне специфічне ураження кісткової тканини, названої Олбрайтом спочатку osteitis fibrosa disseminata. У 1938 році Ліхтенштейн запропонував термін «фіброзна дисплазія», що використовується і сьогодні. Взагалі на початку синдром мав назву «поліосальна фіброзна дисплазія», як це було запропоновано Олбрайтом у 1947 р. [7], за провідним клінічним проявом хвороби, потім назва трансформувалася, і все частіше став використовуватися термін «синдром Олбрайта». Це призвело до того, що в 50–60-х роках XX століття ім’ям Олбрайта назвалися обидва відкриті їм захворювання — хвороба Олбрайта (псевдогіпопаратиреоз) і синдром Олбрайта у випадках поєднання плям гіперпігментації, поліосальної фіброзної дисплазії і передчасного статевого розвитку. У 1962 році в статті з черговим описом клінічного випадку тріади авторами був використаний термін «синдром Мак-Кьюна — Олбрайта» (McCune-Albright syndrome — MAS), і згодом хвороба стала називатися за прізвищами обох авторів [8], а в російській традиції назва захворювання включає прізвища трьох авторів — «синдром Мак-Кьюна — Олбрайта — Брайцева» — МОБ.
Етіологія і патогенез синдрому МОБ
Причина захворювання довгий час залишалася невідомою, хоча припущення про дефект ембріонального розвитку було висловлено ще Ф. Олбрайтом [7]. Істотний внесок у розуміння етіології і патогенезу захворювання зробив німецький дитячий дерматолог Рудольф Хаппл, який виявив у 1986 році мозаїчний характер синдрому. Спостереження дозволили йому зробити висновок про наявність двох клітинних популяцій, що формуються в періоді раннього ембріогенезу внаслідок постзиготної мутації «летального» гена, коли виживання плода можливе тільки в разі поєднання мутантних клітин із клітинами, вільними від мутацій. Це пояснювало поліморфізм проявів, варіабельність локалізації вогнищ фіброзної дисплазії і відсутність спадкових форм. Того ж року зроблено висновок, що в основі захворювання лежить загальний дефект внутрішньоклітинної регуляції синтезу циклічного аденозинмонофосфату (цАМФ). Відкриття ролі Gsα у внутрішньоклітинній передачі сигналу й ідентифікація активуючих мутацій у гені GNAS у 8-му і 9-му екзонах у пацієнтів з об’ємними утвореннями гіпофіза і щитоподібної залози дозволили L.S Weinstein [9] припустити, що «летальним» геном, відповідальним за розвиток захворювання, є ген GNAS, що кодує Gαs [10]. Синдром МОБ викликається соматично активуючими мутаціями в GNAS, розташованому на плечі q хромосоми 20 у положенні 13.2 (20q13.2). Gnas локус є комплексом, що використовує декілька промоторів для отримання різних генних продуктів, у тому числі альфа-субодиниці G секс-стимулюючого білка (Gsα), що частково складається з внутрішнього GTPase-домену, що пов’язує гуаніновий нуклеотид і взаємодіє зі специфічними рецепторами й ефекторами. Власна GTPase-активність Gsα інактивує передачу сигналів рецептора шляхом гідролізу пов’язаного GTP до GDP. Молекулярний дефект і фенотип при синдромі МОБ подані на рис. 1 [1].
/57-1.jpg)
При синдромі МОБ альфа-субодиниця мутує таким чином, щоб викликати конститутивну активацію аденілатциклази і продукувати високі рівні внутрішньоклітинного цАМФ. Це призводить до посилення секреції меланіну, естрадіолу (E2), тесто–стерону (T) [1].
Подальші молекулярно-генетичні дослідження підтвердили ці результати, довівши роль соматичних мутацій у восьмому екзоні гена GNAS R201Н або R201С у розвитку синдрому МОБ і можливість формування інших фенотипів при тій же мутації. Мутації R201 і Q227 у гені GNAS порушують функцію Gsα настільки значимо, що плід здатний вижити лише в разі появи цих мутацій постзиготно [11–13].
При синдромі МОБ спостерігається ураження органів, що походять із різних зародкових листків: епідерміс шкіри, кістки черепа, гіпофіз (ектодерма), кістки осьового і додаткового скелета, гонади, надниркові залози (мезодерма), щитоподібна залоза (ендодерма). Чим раніше виникла мутація, тим більше органів буде уражено. Звідси варіабельність клінічних ознак від легкого синдрому МОБ із мінімумом проявів до тяжких мультикомпонентних форм. Описувана клітинна мутація відбувається на ранніх стадіях ембріогенезу, в перші 10 тижнів вагітності [14]. В цьому випадку мутація локалізується не в усіх клітинах організму, а тільки в окремих клонах мутантної клітини (ця закономірність відома як генетичний мозаїцизм). Отже, чим раніше в процесі ембріонального розвитку відбувається мутація, тим більша кількість клітин матиме структурний дефект, що зумовлює різноманітність клінічних проявів захворювання [15]. Синдром МОБ виникає спорадично, і немає підтверджених випадків вертикальної передачі. Це призвело до недоведеної, але загальноприйнятої моделі патогенезу синдрому МОБ, де постзиготичні мутації GNAS, набуті на початку ембріогенезу, призводять до стану соматичної мозаїки. Відповідно до цієї моделі відсутність вертикальної передачі приблизно відображає ранню летальність у мутаціях зародкової лінії.
Основні клінічні симптоми
Фенотип синдрому МОБ визначається поєднанням факторів, включаючи точку розвитку, в якій відбувається мутація, місця, в які мігрують клітини-мутанти, і специфічні ефекти конститутивних Gsα в уражених тканинах. У випадках, коли залучені тканини, що розвиваються з усіх трьох зародкових шарів (ектодерма, мезодерма й ендодерма), можна припустити, що мутація виникла в одній плюрипотентній клітині до розвитку зародкових шарів.
Головні компоненти синдрому МОБ: фіброзна остеодисплазія; світло-коричнева плямиста пігментація шкіри; передчасний статевий розвиток.
Кістковий симптом. Мозаїцизм проявляється в клінічних особливостях фіброзної дисплазії кісток, у тому числі у вигляді ураження окремих кісток або майже всього скелета [16, 17]. Фіброзні кісти знаходяться, як правило, в довгих трубчастих кістках. Формування великих фіброзних вогнищ призводить до витончення коркового шару, деформації, вкорочення кістки, схильності до спонтанних переломів, особливо часто в проксимальному відділі стегнової кістки, що може перерости в класичну деформацію кульшового суглоба за типом «овеча вовна». Виникають численні вогнища остеопорозу з ділянками остеосклерозу і гіперостозу (особливо в кістках черепа) [18]. Це зумовлює численні переломи з псевдоартрозами, деформації кісткових отворів, що викликає неврологічну симптоматику, біль. Деформація черепа і лицевого скелета може бути значною і супроводжуватися зниженням зору та слуху. Ураження кісток звичайно асиметричне і може бути єдиним симптомом захворювання [19, 20].
Кісткове дозрівання прискорене, але, як правило, стрімкого закриття зон росту не відбувається. Фіброзна дисплазія характеризується складними сайт-специфічними гістологічними змінами, які діляться на три категорії. При класичній формі фіброзна строма від низької до помірно-клітинної форми оточує нерегулярні криволінійні трабекули з плетеної кістки, розташовані у вигляді переривчастого рисунка, зазвичай званого «китайським рисунком листа» [21, 22]. Гістологічно склеротичний тип (пестоїдний) асоціюється з кістками черепа і характеризується щільною склеротичною губчастою кісткою зі складними системами ліній. Склеротичний (гіперцелюлярний) тип асоціюється з гнатичними кістками й основою черепа та містить переривчасті кісткові трабекули, розподілені в упорядкованому, часто паралельному порядку. На всіх ділянках ледь помітні рецидивні ознаки, як правило, допомагають відрізнити фіброзну дисплазію від інших фіброзних уражень, включаючи зіркоподібні остеобласти, волокна Шарпи і надлишок остеоїду, що свідчить про недостатню мінералізацію. При цьому фіброзно-кісткова тканина, як правило, позбавлена гематопоетичних функцій кісткового мозку [23, 24].
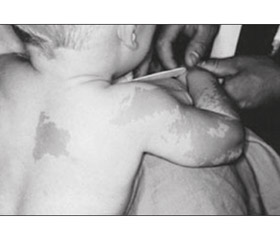
Шкірний симптом. Відразу після народження виникають світло-коричневі плями (кава з молоком — сafé-au-lait) неправильної форми за типом географічної карти або у вигляді «берега Майна» на відміну від плям із рівними краями за типом «берега Каліфорнії» при нейрофіброматозі [25]. Зазвичай плями великих розмірів та частіше розташовуються і більш виражені на стороні ураження, особливо часто — на задній поверхні шиї, спині, стегнах, у поперековій ділянці.
Ендокринна патологія
У дівчаток розвиваються ознаки раннього статевого дозрівання (менструації і вторинні статеві ознаки часто виникають у віці 7 років). Періодичні кісти яєчників призводять до епізодичної секреції естрогену і переривчастих вагінальних кровотеч, що в кінцевому підсумку може призвести до збільшення кісткового віку і зниження зросту [26–33]. Передчасний статевий розвиток при синдромі МОБ починається пізніше і перебігає повільніше, ніж при інших формах передчасного статевого розвитку. Як правило, першим проявом є маткові кровотечі. Вони виникають задовго до настання телархе й адренархе [34, 35]. У хлопчиків статевий розвиток нормальний, у них можуть бути явища гіпергеніталізму або атрофії статевих залоз [36].
У пацієнтів можуть також виявлятися інші ендокринопатії: гіпертиреоз, гіперпаратиреоз, гіперпролактинемія, гіперсоматотропізм, синдром Кушинга. Аномалії щитоподібної залози виникають приблизно у 2/3 пацієнтів, половина з яких пов’язана з вираженим гіпертиреозом [37]. Надлишок гормона росту трапляється рідше, вражаючи 15–20 % пацієнтів [25]. Характерна гіперпродукція фактора росту фібробластів 23 (FGF23) клітинами фіброзної дисплазії, що може призвести до вираженої гіпофосфатемії і рахіту/остеомаляції [38]. Гіперкортицизм — це найрідкісніша ендокринопатія, що пов’язана з синдромом МОБ і виникає виключно в неонатальному періоді через гіперсекрецію кортизолу наднирковими залозами плода [39].
Вікові зміни при синдромі МОБ
Ураження при синдромі МОБ характеризуються віковими гістологічними, рентгенографічними і клінічними змінами. Спостерігалися зниження кількості мутованих BMSC, високий рівень апоптозу в зразках фіброзної дисплазії, менша кількість гістологічних ознак, характерних для хвороби, і в деяких випадках — демонстрація нормальної гістології кісткового мозку, що супроводжується відновленням кровотворення в осіб старшого віку [40]. Це наводить на думку про те, що мутовані скелетні стовбурові клітини в популяції BMSC не здатні до самовідновлення і в кінцевому підсумку поглинаються апоптозом, тоді як співіснуючі нормальні скелетні стовбурові клітини виживають і дозволяють формувати нормальні структури кісткового мозку. У маленьких дітей (< 2 років) часто виявляють гетерогенні ураження на рентгенограмах, які не мають ділянок типу класичного «матового скла». У більш пізньому дитинстві вогнища ураження зазвичай розвивають однорідну рентгенографічну щільність «матового скла», а з віком вони здаються більш щільними і більш склеротичними [41].
Ендокринопатії зазвичай розвиваються в дитинстві або ранньому дитинстві і зберігаються в дорослому житті. Винятки включають неонатальний гіперкортицизм, що може піддаватися спонтанному видужанню після інволюції надниркових залоз плода. FGF23-опосередкована гіпофосфатемія може рости і слабшати разом з активністю захворювання.
Оцінка ендокринопатій є важливим компонентом ведення при синдромі МОБ, оскільки ендокринна дисфункція може посилити захворювання скелета. Зокрема, надлишок гормона росту призводить до макроцефалії і збільшення ризику втрати зору. Гіпофосфатемія пов’язана з підвищеним ризиком переломів і болю в кістках [42–48].
Активуючі мутації GNAS при синдромі МОБ вважаються слабкими онкогенами і можуть вка–зувати на невеликий підвищений ризик злоякісної трансформації в уражених тканинах. Рак, про який повідомляють у зв’язку із синдромом МОБ, вражає кістку, щитоподібну залозу, молочну залозу і яєчко [49, 50]. Прогресування кісткової патології може призвести до тяжкої інвалідизації. Існують відомості про стабілізацію процесу після завершення статевого дозрівання. У зв’язку з цим застосування спеціальної антигонадотропної терапії при цьому захворюванні недоцільне.
Діагностика
Діагноз установлюють на підставі наявності плям кольору кави з молоком, патології кісткової тканини, передчасного статевого розвитку, гіпертиреозу, аномалії надниркових залоз, акромегалії. При лабораторних дослідженнях виявляють підвищення рівня гормонів щитоподібної залози, прищитоподібних та надниркових залоз, а також гормона росту і пролактину. При рентгенологічному дослідженні виявляють фіброзну дисплазію, що уражає декілька кісток. Рентгенологічна картина фіброзної остеодисплазії дуже різноманітна: вогнища розрідження або просвітлення кісткової тканини різних розмірів і форм. Патологічна тканина може розташовуватися у вигляді окремих вогнищ, але можливе й дифузне ураження. При МРТ може виявлятися аденома гіпофіза. При генетичному тестуванні виявляють ген GNAS1.
Клінічний випадок
/59-1.jpg)
Дівчинка віком 2 роки 6 місяців перебувала в хірургічному відділенні з діагнозом: закритий черезвиростковий перелом лівої плечової кістки зі зміщенням. Перелом отримала в результаті падіння на вулиці на розігнуту в ліктьовому суглобі руку. Під час огляду виявлено значний набряк, деформацію лівого ліктьового суглоба, різке обмеження рухів та больовий синдром. Пальці кисті були теплими, рухливими, чутливими, неврологічної симптоматики не встановлено. Налагоджено скелетне витягнення через лівий ліктьовий виросток. На четверту добу після госпіталізації відзначалося збереження зміщення відламків. Виконано хірургічне втручання: остеосинтез лівої плечової кістки (рис. 2). Загоєння первинне, іммобілізація кінцівки гіпсовою пов’язкою відбувалась протягом одного місяця. На контрольному огляді через 1 місяць згинання в ліктьовому суглобі становило 70°, розгинання — 170°. Через 2 місяці в дитини відмічався перелом нижньої третини променевої кістки без зміщення, іммобілізація кінцівки гіпсовою пов’язкою відбулась протягом одного місяця. Ретельний огляд пацієнтки виявив ізольоване телархе, наявність плям кольору кави з молоком у ділянці скроні параорбітально зліва, на грудях із переходом на спину та ліве плече і передпліччя, з нерівними контурами.
/59-2.jpg)
З анамнезу: дитина від першої вагітності. Матері на момент народження дитини було 35 років, батькові — 36 років, хронічних захворювань не мають. Вагітність, зі слів матері, перебігала без особливостей. Маса тіла при народженні становила 2750 г, зріст — 48 см, оцінка за шкалою Апгар — 6/8 балів. На грудному вигодовуванні дитина перебувала до одного року. Мама виявила невеликі пігментовані плями кавового кольору на спині і грудях у віці трьох місяців. У міру зростання дитини плями стали збільшуватися в розмірі, виникали нові плями в скроневій ділянці та ззовні на правому плечі і передпліччі (рис. 2 [51]).
В 1 рік 6 місяців одноразово на підгузку були мізерні кров’яні виділення. Зверталася за місцем проживання до педіатра щодо пігментованих плям, рекомендовано спостереження. У відділенні дівчинка проконсультована дерматологом, дитячим гінекологом, дитячим ендокринологом та генетиком. Призначені додаткові обстеження. Результати: антропометричні показники: зріст — 103,3 см (SDS росту +2,38, що відповідає високорослості), маса — 17,8 кг. Гінекологічний огляд: АХ0 МА1 Pb0 Ме19 міс. Виділення слизові, світлі, мізерні. Зовнішні статеві органи сформовані правильно, клітор не збільшений. Hymen естрогенізований помірно, не гіперемований. Дворучне дослідження: матка розташовується серединно, трохи більша від норми. Придатки з обох сторін не визначаються, пальпаторно ділянка їх локалізації безболісна.
Загальний аналіз крові, сечі без патологічних змін. Гормони крові: фолікулостимулюючий гормон — 0,60 мОД/мл, лютеїнізуючий гормон — 0,20 мОД/мл, естрадіол — 300 пмоль/л, тестостерон — 0,55 нмоль/л, ДГЕА-С — 0,2 мкмоль/л, тиреотропний гормон — 0,5 мМО/л (відзначалося збільшення естрогенів, що є не фізіологічним для даного віку).
При ультразвуковій діагностиці органів малого таза матка розташовується серединно у вигляді тяжа розміром 28 × 7 × 14 мм, М-ехо нечітке, лінійне. Яєчники розташовані низько з боків від матки: правий розміром 16 × 10 мм із фолікулами 2–3 мм, лівий — 14 × 9 мм із фолікулами 2–3 мм.
Рентгенографія кісток рук: виявлена перебудова кісткової тканини в основних і середніх фалангах 2–5-го пальців лівої кисті і меншою мірою — в середніх фалангах 2–5-го пальців правої кисті. Кістковий вік відповідає 3,0–3,5 року, точки окостеніння напівмісячних кісток. Виявлена перебудова структури лівої стегнової кістки за типом фіброзної дисплазії з періостальними нашаруваннями, вогнищевими просвітленнями й елементами остеосклерозу; зазначені зміни виявлялися в дистальній третині вертлюжної западини.
Остеогаммасцинтіграфія — асиметричне накопичення радіофармпрепарату (РФП) у кістках скелета, у ділянці скупчення лівої виличної кістки, лівій половині верхньої і нижньої щелепи, правій великогомілковій кістці, лівій плечовій (незалежні вогнища у верхній і нижній третинах) і правій плечовій (дистальний метафіз) кістках, у куті правої лопатки, у груднині і ребрах. Різко виражена затримка РФП у збиральній системі правої нирки.
Проведена проба з триптореліном — підтверджено гонадотропіннезалежний передчасний статевий розвиток.
Виставлений діагноз: синдром Мак-Кьюна — Олбрайта — Брайцева, передчасний статевий розвиток периферичного генезу, ізосексуальний тип.
Призначено генетичну ДНК-діагностику. Дитина переведена в ендокринологічне відділення.
Висновки
Передчасний статевий розвиток при синдромі МОБ частіше виявляється після першого року життя і характеризується матковою кровотечею і збільшенням грудних залоз (телархе). Адренархе (лобкове й аксилярне оволосіння) не характерне. Основними клінічними ознаками передчасного статевого розвитку в нашому клінічному випадку були збільшення грудних залоз і кров’янисті виділення зі статевих шляхів. Важливо зазначити, що естрогенізація при цьому захворюванні не призводить до настання менархе; властиві швидше менструальноподібні кровотечі, зумовлені різкими коливаннями рівня естрогенів, проте циклічність процесу зазвичай не відзначається. Кістковий вік є важливим критерієм в оцінці хворих із передчасним статевим розвитком. Високий рівень статевих гормонів при тривалому їх впливі значно прискорює прогресування кісткового віку. Кількість плям гіперпігментації корелювала зі ступенем фіброзно-кістозного ураження кісток скелета. Патологічні переломи є класичними ознаками хвороби. Успіх лікування даного захворювання безпосередньо залежить від ранньої діагностики та своєчасного направлення дитини до відповідної ендокринологічної установи. Своєчасні діагностика і лікування сприяють регресу вторинних статевих ознак, запобіганню інвалідизації за рахунок остеодисплазії, і надалі стає можливою реалізація репродуктивної функції.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.