Статтю опубліковано на с.67-70
Введение
Согласно данным ВОЗ, распространенность сахарного диабета в мире к 2025 году составит 299 миллионов заболевших. Однако в 2015 году этот показатель уже превысил прогнозируемый более чем на 100 миллионов человек [1, 2]. Обеспокоенность вызывает прогрессирующий рост числа больных, страдающих от органоспецифических осложнений, обусловленных недостаточным качеством метаболического контроля [3–5]. При всем многообразии факторов, влияющих на изменения метаболических процессов, одним из ведущих остается формирующаяся гипоксия вследствие инсулиновой недостаточности [6–8].
Как предполагают некоторые авторы, в условиях клеточной гипоксии может развиваться энергетический дисбаланс, и наибольший интерес в данном случае представляет изучение изменений, происходящих в митохондриях, так как кислород является одним из основных субстратов для этих клеточных органелл [9–11].
В наше время получены некоторые экспериментальные и клинические доказательства структурно-биохимических дефектов митохондрий в энергозависимых тканях [12, 13].
Но в то же время на фоне огромного количества исследований нет единого представления о закономерностях и функциональном значении изменений ультраструктуры митохондрий при развитии диабетической патологии, обусловленных дефицитом инсулина, и принципы участия митохондрий в них нельзя считать установленными.
Таким образом, целью исследования являлось –изучение динамики изменений в ультраструктурной организации митохондрий миокарда и пульмональной ткани крыс в процессе развития и прогрессирования заболевания на модели экспериментального сахарного диабета (ЭСД).
Материалы и методы
Эксперимент был проведен на 52 белых крысах (самцах) линии Wistar с исходной массой 234,00 ± 2,64 г в возрасте 5–6 месяцев. Работу с животными осуществляли согласно положениям «Общих этических принципов экспериментов на животных», принятых I Национальным конгрессом по биоэтике (Киев, 2001), и международным требованиям «Европейской конвенции защиты позвоночных животных, которые используются в экспериментальных и других научных целях».
Животные были разделены на следующие группы. І группу составили 25 лабораторных крыс линии Wistar с массой тела 230,70 ± 3,16 г. У подопытных животных данной группы нами был смоделирован стрептозотоциновый сахарный диабет путем однократного внутрибрюшного введения стрептозотоцина (Sigma, США) в 0,1 М цитратном буфере рН 4,5 в дозе 60 мг/кг. Введение стрептозотоцина осуществляли после предварительной 24-часовой депривации пищи при сохраненном доступе к воде. С целью формирования полного и стабильного диабета животных содержали на протяжении 11 суток на стандартной диете. Определение глюкозы крови из хвостовой вены проводили глюкозооксидазным методом. Для дальнейшего исследования использовали только особей с повышенным уровнем глюкозы (> 11 ммоль/л).
ІІ группу составили подопытные крысы (27 особей) линии Wistar с массой тела 228,30 ± 1,12 г.
Учитывая стойкие цифры тощаковой гликемии во ІІ группе на уровне 25,7 ± 1,4 ммоль/л и прогрессирующее снижение массы тела подопытных грызунов до 186,0 ± 1,8 г без получения сахароснижающей терапии, на 18-й неделе животных выводили из эксперимента путем декапитации после предварительной 30-минутной анестезии с помощью ингаляции паров севофлурана.
Фиксацию материала производили немедленно, внося образцы ткани в забуференный 2,5% раствор глутарового альдегида. Дофиксация материала осуществлялась с помощью реактива Колфилда (на основе 2% раствора четырехокиси осмия, рН 7,3) (реактивы фирмы Sigma, США). Обезвоживание материала производили в спиртах возрастающей концентрации, абсолютном спирте и ацетоне. Последующая заливка в эпонаралдит (фирмы Fluka, Швейцария) проводилась по общепринятой методике [14].
Ультратонкие срезы толщиной 40–60 нм для просмотра в электронном микроскопе контрастировали 1% раствором уранилацетата и раствором цитрата свинца (реактивы фирмы Sigma, США) по методике Рейнольдса [15].
Просмотр препаратов осуществляли с помощью электронного микроскопа ПЕМ-125К (Украина).
Результаты и обсуждение
В результате проведенного исследования у лабораторных животных с ЭСД были зафиксированы идентичные ультраструктурные изменения митохондрий в кардио- и альвеолоцитах в виде фрагментации мембран, что, с одной стороны, может свидетельствовать о снижении эффективности работы митохондрий и энергопотенциала, создаваемого на этих мембранах на фоне стойкой декомпенсации углеводного обмена, а с другой — характеризоваться как защитный ответ клеток, направленный на предотвращение их гибели, что согласуется с данными других авторов [16, 17].
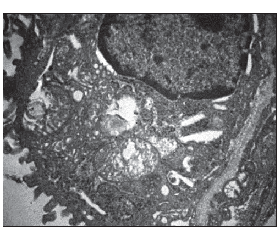
При дальнейшем анализе в диабетической группе были определены признаки разрушения крист и вакуолизация митохондрий в исследуемых тканях, что подразумевало снижение их функциональной активности и являлось первичным признаком аутолиза клеток при не контролируемой противодиабетическими препаратами гипергликемии в отличие от митохондрий крыс без сахарного диабета (рис. 1).
Следует отметить, что дезорганизация митохондрий наблюдалась в виде х-конденсации и набухания с возрастанием величины их среднего диаметра как в миокарде, так и в изучаемой легочной ткани по сравнению с группой недиабетических крыс (рис. 2, 3).
Именно конденсация и набухание митохондрий, согласно современным представлениям, свидетельствуют о функциональном напряжении клетки вследствие нарастающего кислородного голодания и развития гипоксических процессов в тканях [18, 19].
Стоит отметить, что в ряде препаратов альвеолярной ткани при стрептозотоциновом диабете помимо наличия структурно поврежденных митохондрий определялось отсутствие активации их морфогенеза (рис. 4).
В кардиомиоцитах диабетических крыс также были верифицированы начальные стадии проявлений аутофагии митохондрий с уплотнением митохондриальных мембран и повышением электронной плотности отдельных пространств между кристами (риc. 5).
Выводы
1. В результате проведенного исследования нами получены убедительные доказательства ультраструктурных изменений митохондрий в виде –дезорганизации и редукции их структур под влиянием инсулиновой недостаточности без сахароснижающего лечения.
2. В условиях экспериментального диабета состояние изучаемых клеточных органелл соответствовало условиям сниженной выработки АТФ, что ограничивало их энергетическую функцию или делало митохондрии в изучаемых тканях энергетически несостоятельными.
3. Полученные нами данные свидетельствовали о том, что при стойкой диабетической декомпенсации наблюдаются нарушения внутриклеточной биоэнергетики, что вызывало нарушения биосинтетических и репаративных процессов на субклеточном уровне и указывало на развитие вторичной митохондриальной недостаточности в миокарде и тканях дыхательной системы.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Guariguata L. Contribute data to the 6th edition of the IDF Diabetes Atlas // Diabetes Res. Clin. Pract. — 2013. — Vol. 100 (2). — P. 280-281.
2. Twig G., Yaniv G., Levine H. et al. Body-Mass Indexin 2.3 Million Adolescents and Cardiovascular Deathin Adulthood // N. Engl. J. Med. — 2016. — Vol. 374 (25). — P. 2430-2440.
3. Danaei G., Finucane M.M., Lu Y. et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants // Lancet. — 2011. — Vol. 378 (9785). — P. 31-40.
4. Dosluoglu H.H., Lall P., Nader N.D. et al. Insulin use is associated with poor limb salvage and survival in diabetic patients with chronic limb ischemia // J. Vasc. Surg. — 2010. — Vol. 51 (5). — P. 1178-1189.
5. Zhou B., Lu Y., Hajifathalian K. et al. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants // Lancet. — 2016. — Vol. 387 (10027). — P. 1513-1530.
6. Wheatley C.M., Baldi J.C., Cassuto N.A. et al. Glycemic control influences lung membrane diffusion and oxygen saturation in exercise-trained subjects with type 1 diabetes: alveolar-ca–pillary membrane conductance in type 1 diabetes // Eur. J. Appl. Physiol. — 2011. — Vol. 111 (3).— P. 567-578.
7. Mercer J.R., Liu H. The Evaluation of Hypoxiain Diabetes // Developments in Nuclear Medicine. — 1999. — Vol. 33. — P. 129-153.
8. Gomez-Valdes A. Chronic Hypoxia Causes Disorder of Glucose Metabolism and a Specific Type of Diabetes // Journal of Endocrinology and Diabetes Mellitus. — Vol. 2 (issue 2). — P. 53-57.
9. Armstrong J.S., Whiteman M. Measurement of reactive oxygen species in cells and mitochondria // Methods Cell. Biol. — 2007. — Vol. 80. — P. 355-377.
10. Roth M., Black J.L. An imbalance in C/EBPs and increased mitochondrial activity in asthmatic airway smooth muscle cells: novel targets in asthma therapy? // Br. J. Pharmacol. — 2009. — Vol. 157 (3). — P. 334-341.
11. Disorders of Mitochondrial Energy Metabolism / Rahman S., Janssen C.H.M. — 2016. // Oxford Medicine Online: http://dx.doi.org/10.1093/med/9780199972135.003.0007
12. Mayevsky A., Rogatsky G.G. Mitochondrial function in vivo evaluated by NADH fluorescence: from animal models to human studies // Am. J. Physiol. Cell. Physiol. — 2007. — Vol. 292 (2). — Р. 615-640.
13. Schaible N., Delmotte P., Sieck G.C.Mitochondrial Excitation-Energy Coupling in Airway Smooth Muscle // Respiratory Medi–cine. — 2014. http://dx.doi.org/10.1007/978-1-4939-0829-5_5.
14. Кaрупу В.Я. Элeктрoннaя микрocкoпия / В.Я. Кaрупу. — К.: Вищa шкoлa, 1984. — 208 c.
15. Вeйбeль Э.Р. Мoрфoмeтрия лeгких чeлoвeкa / Э.Р. Вeйбeль: Пeр. c aнгл. — М.: Мeдицинa, 1970. — 170 c.
16. Carraro M., Bernardi P. Calcium andreactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast // Cell. Calcium. — 2016. — Vol. 60 (2). — Р. 102-107.
17. Carraro M., Giorgio V., Šileikytė J. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition // J. Biol. Chem. — 2014. — Vol. 289 (23). — 15980-15985.
18. Brooks G.A. Energy Flux, Lactate Shuttling, Mitochondrial Dynamics, and Hypoxia // Adv. Exp. Med. Biol. — 2016. — Vol. 903. — 439-455.
19. Gonchar O., Mankovskaya I. Effect of moderate hypoxia/reoxygenation on mitochondrial adaptation to acute severe hypoxia // Acta Biol. Hung. — 2009. — Vol. 60 (2). — Р. 185-194.

/68-1.gif)
/68-2.gif)
/69-1.gif)
/69-2.gif)